
JM; Sousa, Bizao
Elastic Properties of Graphyne-based Nanotubes Online
2019, (ArXiv preprint.).
@online{deSousa2019b,
title = {Elastic Properties of Graphyne-based Nanotubes},
author = {de Sousa, JM; , Bizao, RA; Sousa Filho, VP; Aguiar, AL; Coluci, VR; Pugno, NM; Girao, EC; Souza Filho, AG; Galvao, DS},
url = {https://arxiv.org/pdf/1905.02104.pdf},
year = {2019},
date = {2019-04-07},
abstract = {Graphyne nanotubes (GNTs) are nanostructures obtained from rolled up graphyne sheets,
in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes
are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to
conventional CNTs, GNTs can present different chiralities and electronic properties. Because
of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their
mechanical properties. In this work, we studied the mechanical response of GNTs under
tensile stress using fully atomistic molecular dynamics simulations and density functional
theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs
at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller
ultimate strength and Young’s modulus values. This is a consequence of the combined
effects of the existence of triple bonds and increased porosity/flexibility due to the presence
of acetylenic groups.},
note = {ArXiv preprint.},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes
are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to
conventional CNTs, GNTs can present different chiralities and electronic properties. Because
of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their
mechanical properties. In this work, we studied the mechanical response of GNTs under
tensile stress using fully atomistic molecular dynamics simulations and density functional
theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs
at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller
ultimate strength and Young’s modulus values. This is a consequence of the combined
effects of the existence of triple bonds and increased porosity/flexibility due to the presence
of acetylenic groups.
JM; Sousa, Bizao
Elastic Properties of Graphyne-Based Nanotubes Journal Article
In: Computational Materials Science, vol. 170, pp. 109153, 2019.
@article{deSousa2019c,
title = {Elastic Properties of Graphyne-Based Nanotubes},
author = {de Sousa, JM; , Bizao, RA; Sousa Filho, VP; Aguiar, AL; Coluci, VR; Pugno, NM; Girao, EC; Souza Filho, AG; Galvao, DS},
url = {https://www.sciencedirect.com/science/article/pii/S0927025619304525?dgcid=coauthor#s0040},
doi = {10.1016/j.commatsci.2019.109153},
year = {2019},
date = {2019-04-03},
journal = {Computational Materials Science},
volume = {170},
pages = {109153},
abstract = {Graphyne nanotubes (GNTs) are nanostructures obtained from rolled up graphyne sheets, in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to conventional CNTs, GNTs can present different chiralities and electronic properties. Because of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their mechanical properties. In this work, we studied the mechanical response of GNTs under tensile stress using fully atomistic molecular dynamics simulations and density functional theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller ultimate strength and Young’s modulus values. This is a consequence of the combined effects of the existence of triple bonds and increased porosity/flexibility due to the presence of acetylenic groups.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Solis, Daniel; Damasceno Borges, Daiane; Woellner, Cristiano; Galvao, Douglas
Structural and Thermal Stability of Graphyne and Graphdiyne Nanoscroll Structures (invited paper) Journal Article
In: ACS Applied Materials and Interfaces, vol. 11, pp. 2670−2676, 2019.
@article{Solis2019,
title = {Structural and Thermal Stability of Graphyne and Graphdiyne Nanoscroll Structures (invited paper)},
author = {Solis, Daniel and Damasceno Borges, Daiane and Woellner, Cristiano and Galvao,
Douglas},
url = {https://pubs.acs.org/doi/10.1021/acsami.8b03481},
doi = {10.1021/acsami.8b03481},
year = {2019},
date = {2019-01-23},
journal = {ACS Applied Materials and Interfaces},
volume = {11},
pages = {2670−2676},
abstract = {Graphynes and graphdiynes are generic names for families of two-dimensional carbon allotropes, where acetylenic groups connect benzenoid-like hexagonal rings, with the coexistence of sp and sp2 hybridized carbon atoms. The main differences between graphynes and graphdiynes are the number of acetylenic groups (one and two for graphynes and graphdiynes, respectively). Similarly to graphene nanoscrolls, graphyne and graphdiynes nanoscrolls are nanosized membranes rolled into papyrus-like structures. In this work we studied through molecular dynamics simulations, using reactive potentials, the structural and thermal (up to 1000 K) stability of α,β,γ-graphyne and α,β,γ-graphdiyne scrolls. Our results demonstrate that stable nanoscrolls can be created for all the structures studied here, although they are less stable than corresponding graphene scrolls. This can be elucidated as a result of the higher graphyne/graphdiyne structural porosity in relation to graphene, and as a consequence, the π–π stacking interactions decrease.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Solis, Daniel; Borges, Daiane D.; Woellner, Cristiano F.; Galvao, Douglas S.
Structural and Thermal Stability of Graphyne and Graphdiyne Nanoscroll Structures Online
2018, visited: 02.03.2018, (preprint ArXiv: 1803.00154).
@online{Solis2018b,
title = {Structural and Thermal Stability of Graphyne and Graphdiyne Nanoscroll Structures},
author = {Daniel Solis and Daiane D. Borges and Cristiano F. Woellner and Douglas S. Galvao},
url = {https://arxiv.org/abs/1803.00154},
year = {2018},
date = {2018-03-02},
urldate = {2018-03-02},
abstract = {Graphynes and graphdiynes are generic names for families of two-dimensional carbon allotropes,
where acetylenic groups connect benzenoid-like hexagonal rings, with the co-existence of sp and
sp
2 hybridized carbon atoms. The main differences between graphynes and graphdiynes are the
number of acetylenic groups (one and two for graphynes and graphdiynes, respectively).
Similarly to graphene nanoscrolls, graphyne and graphdiynes nanoscrolls are nanosized
membranes rolled up into papyrus-like structures. In this work we investigated through fully
atomistic reactive molecular dynamics simulations the structural and thermal (up to 1000K)
stability of α,β,γ-graphyne and α,β,γ-graphdiyne scrolls. Our results show that stable nanoscrolls
can be formed for all the structures investigated here, although they are less stable than
corresponding graphene scrolls. This can be explained as a consequence of the higher
graphyne/graphdiyne structural porosity in relation to graphene, which results in decreased π-π
stacking interactions. },
note = {preprint ArXiv: 1803.00154},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
where acetylenic groups connect benzenoid-like hexagonal rings, with the co-existence of sp and
sp
2 hybridized carbon atoms. The main differences between graphynes and graphdiynes are the
number of acetylenic groups (one and two for graphynes and graphdiynes, respectively).
Similarly to graphene nanoscrolls, graphyne and graphdiynes nanoscrolls are nanosized
membranes rolled up into papyrus-like structures. In this work we investigated through fully
atomistic reactive molecular dynamics simulations the structural and thermal (up to 1000K)
stability of α,β,γ-graphyne and α,β,γ-graphdiyne scrolls. Our results show that stable nanoscrolls
can be formed for all the structures investigated here, although they are less stable than
corresponding graphene scrolls. This can be explained as a consequence of the higher
graphyne/graphdiyne structural porosity in relation to graphene, which results in decreased π-π
stacking interactions.
G. Brunetto J.M. de Sousa, V. R. Coluci
Torsional “superplasticity” of graphyne nanotubes Journal Article
In: Carbon, vol. 96, pp. 14-19, 2016.
@article{deSousa2016,
title = {Torsional “superplasticity” of graphyne nanotubes},
author = {J.M. de Sousa, G. Brunetto, V.R. Coluci, D.S. Galvao },
url = {http://www.sciencedirect.com/science/article/pii/S000862231530258X},
doi = { http://dx.doi.org/10.1016/j.carbon.2015.09.039},
year = {2016},
date = {2016-01-01},
journal = {Carbon},
volume = {96},
pages = {14-19},
abstract = {Graphyne is a planar two-dimensional carbon allotrope formed by atoms in sp, sp2, and sp3 hybridized states. Topologically graphyne nanotubes (GNTs) can be considered as cylindrically rolled up graphyne sheets, similarly as carbon nanotubes (CNTs) can be considered rolled up graphene sheets. Due to the presence of single, double, and triple bonds, GNTs exhibit porous sidewalls that can be exploited in many diverse applications. In this work, we investigated the mechanical behavior of GNTs under torsional strains through reactive molecular dynamics simulations. Our results show that GNTs are more flexible than CNTs and exhibit “superplasticit”, with fracture angles that are up to 35 times higher than the ones reported to CNTs. This GNT “superplastic” behavior can be explained in terms of irreversible recon- struction processes (mainly associated with the triple bonds) that occur during torsional strains.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Gustavo Brunetto Jose M. de Sousa, Vitor R. Coluci
Torsional "Superplasticity" of Graphyne Nanotubes Online
2015, (ArXiv reprint of Torsional "Superplasticity" of Graphyne Nanotubes, published in Carbon 96, 14 (2016).).
@online{deSousa2015b,
title = {Torsional "Superplasticity" of Graphyne Nanotubes},
author = {Jose M. de Sousa, Gustavo Brunetto, Vitor R. Coluci, Douglas S. Galvao},
url = {http://arxiv.org/abs/1509.08746},
year = {2015},
date = {2015-09-29},
abstract = {Graphyne is a planar two-dimensional carbon allotrope formed by atoms in sp, sp2, and sp3 hybridized states. Topologically graphyne nanotubes (GNTs) can be considered as cylindrically rolled up graphyne sheets, similarly as carbon nanotubes (CNTs) can be considered rolled up graphene sheets. Due to the presence of single, double, and triple bonds, GNTs exhibit porous sidewalls that can be exploited in many diverse applications. In this work, we investigated the mechanical behavior of GNTs under torsional strains through reactive molecular dynamics simulations. Our results show that GNTs are more flexible than CNTs and exhibit 'superplasticity', with fracture angles that are up to 35 times higher than the ones reported to CNTs. This GNT 'superplastic' behavior can be explained in terms of irreversible reconstruction processes (mainly associated with the triple bonds) that occur during torsional strains.},
note = {ArXiv reprint of Torsional "Superplasticity" of Graphyne Nanotubes, published in Carbon 96, 14 (2016).},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Pedro A. S. Autreto, Douglas S. Galvao
Site dependent hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation Online
2015, (ArXiv draft of MRS Proceedings, 1726, mrsf14-1726-j02-02 (2015)).
@online{Autreto2015,
title = {Site dependent hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Pedro A. S. Autreto, Douglas S. Galvao},
url = {http://arxiv.org/abs/1501.04521},
year = {2015},
date = {2015-01-19},
journal = {arXiv preprint 1501.04521},
abstract = {Graphyne is a generic name for a carbon allotrope family of 2D structures, where acetylenic groups connect benzenoid rings, with the coexistence of sp and sp2 hybridized carbon atoms. In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics and structural changes of the hydrogenation of ALPHA, BETA, GAMMA graphyne forms. Our results showed that the existence of different sites for hydrogen bonding, related to single and triple bonds, makes the process of incorporating hydrogen atoms into graphyne membranes much more complex than the graphene ones. Our results also show that hydrogenation reactions are strongly site dependent and that the sp-hybridized carbon atoms are the preferential sites to chemical attacks. In our cases, the effectiveness of the hydrogenation (estimated from the number of hydrogen atoms covalently bonded to carbon atoms) follows the ALPHA, BETA, GAMMA graphyne structure ordering.},
note = {ArXiv draft of MRS Proceedings, 1726, mrsf14-1726-j02-02 (2015)},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Pedro A. S. Autreto, Douglas S. Galvao
Site Dependent Hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation Proceedings
vol. 1726, no. mrsf14-1726-j02-02, 2015, (MRS Proceedings, 1726, mrsf14-1726-j02-02 ).
@proceedings{Autreto2015b,
title = {Site Dependent Hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Pedro A. S. Autreto, Douglas S. Galvao},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=9702693&fulltextType=RA&fileId=S1946427415004649},
doi = {10.1557/opl.2015.464},
year = {2015},
date = {2015-01-01},
journal = {Mater. Res. Soc. Symp. Proc. },
volume = {1726},
number = {mrsf14-1726-j02-02},
abstract = {Graphyne is a generic name for a carbon allotrope family of 2D structures, where acetylenic groups connect benzenoid rings, with the coexistence of sp and sp2 hybridized carbon atoms. In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics and structural changes of the hydrogenation of α, β, and γ graphyne forms. Our results showed that the existence of different sites for hydrogen bonding, related to single and triple bonds, makes the process of incorporating hydrogen atoms into graphyne membranes much more complex than the graphene ones. Our results also show that hydrogenation reactions are strongly site dependent and that the sp-hybridized carbon atoms are the preferential sites to chemical attacks. In our cases, the effectiveness of the hydrogenation (estimated from the number of hydrogen atoms covalently bonded to carbon atoms) follows the α, β, γ-graphyne structure ordering.},
note = {MRS Proceedings, 1726, mrsf14-1726-j02-02 },
keywords = {},
pubstate = {published},
tppubtype = {proceedings}
}
Autreto, PAS; de Sousa, JM; Galvao, DS
Site-dependent hydrogenation on graphdiyne Journal Article
In: Carbon, vol. 77, pp. 829–834, 2014.
@article{autreto2014site,
title = {Site-dependent hydrogenation on graphdiyne},
author = {Autreto, PAS and de Sousa, JM and Galvao, DS},
url = {http://www.sciencedirect.com/science/article/pii/S0008622314005429},
year = {2014},
date = {2014-01-01},
journal = {Carbon},
volume = {77},
pages = {829--834},
publisher = {Pergamon},
abstract = {Graphene is one of the most important materials in science today due to its unique and remarkable electronic, thermal and mechanical properties. However in its pristine state, graphene is a gapless semiconductor, what limits its use in transistor electronics. In part due to the revolution created by graphene in materials science, there is a renewed interest in other possible graphene-like two-dimensional structures. Examples of these structures are graphynes and graphdiynes, which are two-dimensional structures, composed of carbon atoms in sp2 and sp-hybridized states. Graphdiynes (benzenoid rings connecting two acetylenic groups) were recently synthesized and some of them are intrinsically nonzero gap systems. These systems can be easily hydrogenated and the relative level of hydrogenation can be used to tune the band gap values. We have investigated, using fully reactive molecular dynamics (ReaxFF), the structural and dynamics aspects of the hydrogenation mechanisms of graphdiyne membranes. Our results showed that the hydrogen bindings have different atom incorporation rates and that the hydrogenation patterns change in time in a very complex way. The formation of correlated domains reported to hydrogenated graphene is no longer observed in graphdiyne cases.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Autreto, PA; de Sousa, JM; Galvao, DS
On the Dynamics of Graphdiyne Hydrogenation Proceedings
Cambridge University Press, vol. 1549, 2013.
@proceedings{autreto2013dynamics,
title = {On the Dynamics of Graphdiyne Hydrogenation},
author = {Autreto, PA and de Sousa, JM and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8915680&fileId=S1946427413006088},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1549},
pages = {59--64},
publisher = {Cambridge University Press},
abstract = {Graphene is a two-dimensional (2D) hexagonal array of carbon atoms in sp2-hybridized states. Graphene presents unique and exceptional electronic, thermal and mechanical properties. However, in its pristine state graphene is a gapless semiconductor, which poses some limitations to its use in some transistor electronics. Because of this there is a renewed interest in other possible two-dimensional carbon-based structures similar to graphene. Examples of this are graphynes and graphdiynes, which are two-dimensional structures, composed of carbon atoms in sp2 and sp-hybridized states. Graphdiynes (benzenoid rings connecting two acetylenic groups) were recently synthesized and they can be intrinsically nonzero gap systems. These systems can be easily hydrogenated and the amount of hydrogenation can be used to tune the band gap value. In this work we have investigated, through fully atomistic molecular dynamics simulations with reactive force field (ReaxFF), the structural and dynamics aspects of the hydrogenation mechanisms of graphdiyne membranes. Our results showed that depending on whether the atoms are in the benzenoid rings or as part of the acetylenic groups, the rates of hydrogenation are quite distinct and change in time in a very complex pattern. Initially, the most probable sites to be hydrogenated are the carbon atoms forming the triple bonds, as expected. But as the amount of hydrogenation increases in time this changes and then the carbon atoms forming single bonds become the preferential sites. The formation of correlated domains observed in hydrogenated graphene is no longer observed in the case of graphdiynes. We have also carried out ab initio DFT calculations for model structures in order to test the reliability of ReaxFF calculations.},
keywords = {},
pubstate = {published},
tppubtype = {proceedings}
}
Coluci, VR; Braga, SF; Legoas, SB; Galvao, DS; Baughman, RH
New families of carbon nanotubes based on graphyne motifs Journal Article
In: Nanotechnology, vol. 15, no. 4, pp. S142, 2004.
@article{coluci2004new,
title = {New families of carbon nanotubes based on graphyne motifs},
author = {Coluci, VR and Braga, SF and Legoas, SB and Galvao, DS and Baughman, RH},
url = {http://iopscience.iop.org/0957-4484/15/4/006},
year = {2004},
date = {2004-01-01},
journal = {Nanotechnology},
volume = {15},
number = {4},
pages = {S142},
publisher = {IOP Publishing},
abstract = {Electronic properties of proposed new families of carbon single walled nanotubes are investigated. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Analogous to ordinary nanotubes, armchair, zigzag and chiral graphyne nanotubes are possible. Tight-binding and ab initio density functional methods were used to predict the electronic properties of these unusual nanotubes. Of the three graphyne nanotube families analysed here, two provide metallic behaviour for armchair tubes and either metallic or semiconducting behaviour for zigzag nanotubes. For the other graphyne nanotube family investigated a diameter and chirality independent bandgap is predicted and a bandgap modulation study by structural distortions has been carried out for small longitudinal tube deformations. Interestingly, while the bandgap is insensitive to structure, the stress-induced bandgap changes can strongly depend both on the nanotube type and whether the strain is tensile or compressive.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Coluci, VR; Galvao, DS; Baughman, RH
Theoretical investigation of electromechanical effects for graphyne carbon nanotubes Journal Article
In: The Journal of chemical physics, vol. 121, no. 7, pp. 3228–3237, 2004.
@article{coluci2004theoretical,
title = {Theoretical investigation of electromechanical effects for graphyne carbon nanotubes},
author = {Coluci, VR and Galvao, DS and Baughman, RH},
url = {http://scitation.aip.org/content/aip/journal/jcp/121/7/10.1063/1.1772756},
year = {2004},
date = {2004-01-01},
journal = {The Journal of chemical physics},
volume = {121},
number = {7},
pages = {3228--3237},
publisher = {AIP Publishing},
abstract = {We present a theoretical study of the electronic and mechanical properties of graphyne-based nanotubes (GNTs). These semiconducting nanotubes result from the elongation of one-third of the covalent interconnections of graphite-based nanotubes by the introduction of yne groups. The effect of charge injection on the dimensions of GNTs was investigated using tight-binding calculations. Low amounts of electron injection are predicted to cause qualitatively different responses for armchair and zigzag graphyne nanotubes. Although the behavior is qualitatively similar to the usual carbon nanotubes, the charge-induced strains are predicted to be smaller for the GNTs than for ordinary single walled carbon nanotubes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Coluci, VR; Braga, SF; Legoas, SB; Galvao, DS; Baughman, RH
Families of carbon nanotubes: Graphyne-based nanotubes Journal Article
In: Physical Review B, vol. 68, no. 3, pp. 035430, 2003.
@article{coluci2003families,
title = {Families of carbon nanotubes: Graphyne-based nanotubes},
author = {Coluci, VR and Braga, SF and Legoas, SB and Galvao, DS and Baughman, RH},
url = {http://journals.aps.org/prb/abstract/10.1103/PhysRevB.68.035430},
year = {2003},
date = {2003-01-01},
journal = {Physical Review B},
volume = {68},
number = {3},
pages = {035430},
publisher = {APS},
abstract = {New families of carbon single-walled nanotubes are proposed and their electronic structures are investigated. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Analogously to ordinary nanotubes, armchair, zigzag, and chiral graphyne nanotubes are possible. We here predict the electronic properties of these unusual nanotubes using tight-binding and ab initio density functional methods. Of the three graphyne nanotube families analyzed here, two provide metallic behavior for armchair tubes and either metallic or semiconducting behavior for zigzag nanotubes. A diameter- and chirality-independent band gap is predicted for the other investigated graphyne family, as well as an oscillatory dependence of the effective mass on nanotube diameter.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Coluci, VR; Braga, SF; Legoas, SB; Galvao, DS; Baughman, RH
Graphyne Nanotubes: New Families of Carbon Nanotubes Proceedings
Warrendale, Pa.; Materials Research Society; 1999, vol. 739, 2003.
@proceedings{coluci2003graphyne,
title = {Graphyne Nanotubes: New Families of Carbon Nanotubes},
author = {Coluci, VR and Braga, SF and Legoas, SB and Galvao, DS and Baughman, RH},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8031794},
year = {2003},
date = {2003-01-01},
journal = {MATERIALS RESEARCH SOCIETY SYMPOSIUM PROCEEDINGS},
volume = {739},
pages = {175--180},
publisher = {Warrendale, Pa.; Materials Research Society; 1999},
abstract = {Fundamentally new families of carbon single walled nanotubes are proposed. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Similarly to ordinary nanotubes, armchair, zig-zag, and chiral graphyne nanotubes are possible. We present here results for the electronic properties of graphyne based tubes obtained from tight-binding and ab initio density functional methods.},
keywords = {},
pubstate = {published},
tppubtype = {proceedings}
}
Coluci, V R; Braga, SF; Legoas, Sergio B; Galvao, Douglas S; Baughman, RH
New families of carbon nanotubes Journal Article
In: arXiv preprint cond-mat/0207085, 2002.
@article{coluci2002new,
title = {New families of carbon nanotubes},
author = {Coluci, V R and Braga, SF and Legoas, Sergio B and Galvao, Douglas S and Baughman, RH},
url = {http://arxiv.org/abs/cond-mat/0207085},
year = {2002},
date = {2002-01-01},
journal = {arXiv preprint cond-mat/0207085},
abstract = {Fundamentally new families of carbon single walled nanotubes are proposed. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Similarly to ordinary nanotubes, arm-chair, zig-zag, and chiral graphyne nanotubes are possible. Electronic properties, predicted using tight-binding and ab initio density functional methods, show a rich variety of metallic and semiconducting behaviors.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2019
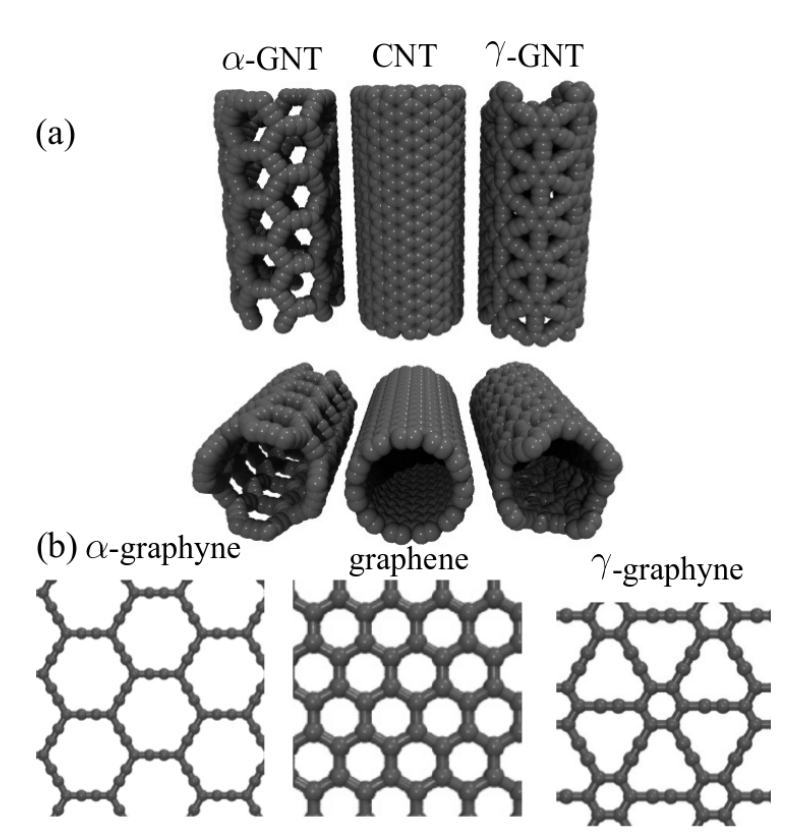
JM; Sousa, Bizao
Elastic Properties of Graphyne-based Nanotubes Online
2019, (ArXiv preprint.).
Abstract | Links | BibTeX | Tags: DFT, Graphynes, Molecular Dynamics, Nanotubes
@online{deSousa2019b,
title = {Elastic Properties of Graphyne-based Nanotubes},
author = {de Sousa, JM; , Bizao, RA; Sousa Filho, VP; Aguiar, AL; Coluci, VR; Pugno, NM; Girao, EC; Souza Filho, AG; Galvao, DS},
url = {https://arxiv.org/pdf/1905.02104.pdf},
year = {2019},
date = {2019-04-07},
abstract = {Graphyne nanotubes (GNTs) are nanostructures obtained from rolled up graphyne sheets,
in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes
are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to
conventional CNTs, GNTs can present different chiralities and electronic properties. Because
of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their
mechanical properties. In this work, we studied the mechanical response of GNTs under
tensile stress using fully atomistic molecular dynamics simulations and density functional
theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs
at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller
ultimate strength and Young’s modulus values. This is a consequence of the combined
effects of the existence of triple bonds and increased porosity/flexibility due to the presence
of acetylenic groups.},
note = {ArXiv preprint.},
keywords = {DFT, Graphynes, Molecular Dynamics, Nanotubes},
pubstate = {published},
tppubtype = {online}
}
in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes
are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to
conventional CNTs, GNTs can present different chiralities and electronic properties. Because
of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their
mechanical properties. In this work, we studied the mechanical response of GNTs under
tensile stress using fully atomistic molecular dynamics simulations and density functional
theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs
at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller
ultimate strength and Young’s modulus values. This is a consequence of the combined
effects of the existence of triple bonds and increased porosity/flexibility due to the presence
of acetylenic groups.
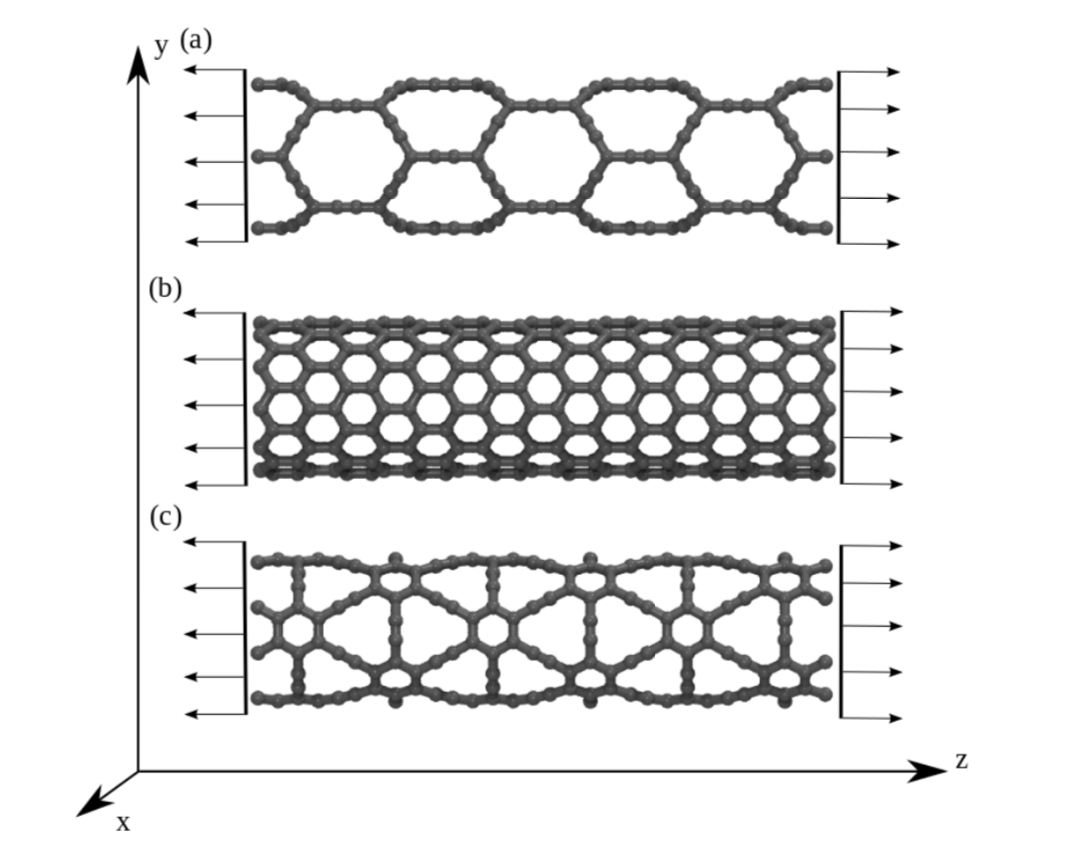
JM; Sousa, Bizao
Elastic Properties of Graphyne-Based Nanotubes Journal Article
In: Computational Materials Science, vol. 170, pp. 109153, 2019.
Abstract | Links | BibTeX | Tags: DFT, Graphynes, Molecular Dynamics, Nanotubes
@article{deSousa2019c,
title = {Elastic Properties of Graphyne-Based Nanotubes},
author = {de Sousa, JM; , Bizao, RA; Sousa Filho, VP; Aguiar, AL; Coluci, VR; Pugno, NM; Girao, EC; Souza Filho, AG; Galvao, DS},
url = {https://www.sciencedirect.com/science/article/pii/S0927025619304525?dgcid=coauthor#s0040},
doi = {10.1016/j.commatsci.2019.109153},
year = {2019},
date = {2019-04-03},
journal = {Computational Materials Science},
volume = {170},
pages = {109153},
abstract = {Graphyne nanotubes (GNTs) are nanostructures obtained from rolled up graphyne sheets, in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to conventional CNTs, GNTs can present different chiralities and electronic properties. Because of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their mechanical properties. In this work, we studied the mechanical response of GNTs under tensile stress using fully atomistic molecular dynamics simulations and density functional theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller ultimate strength and Young’s modulus values. This is a consequence of the combined effects of the existence of triple bonds and increased porosity/flexibility due to the presence of acetylenic groups.},
keywords = {DFT, Graphynes, Molecular Dynamics, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
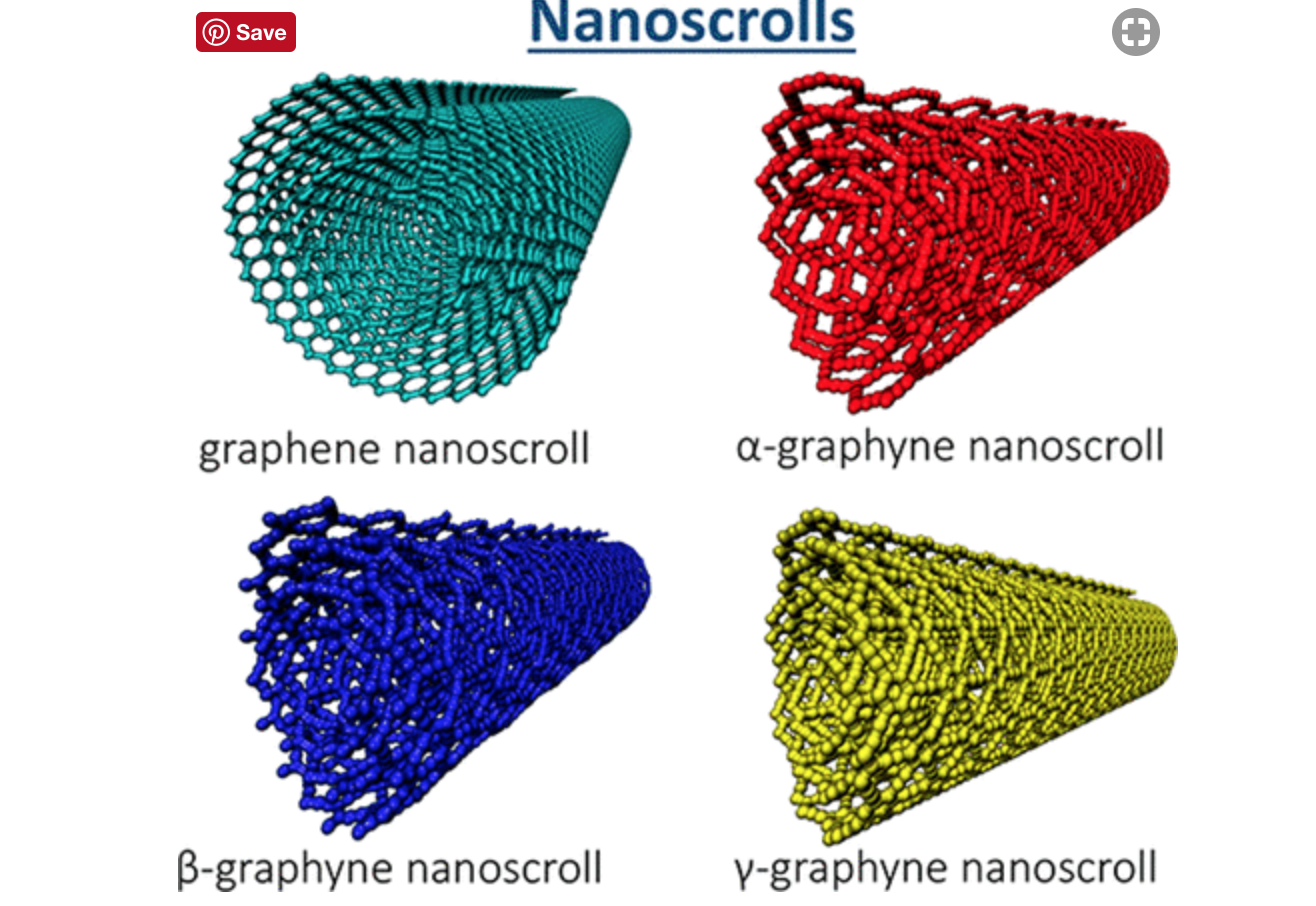
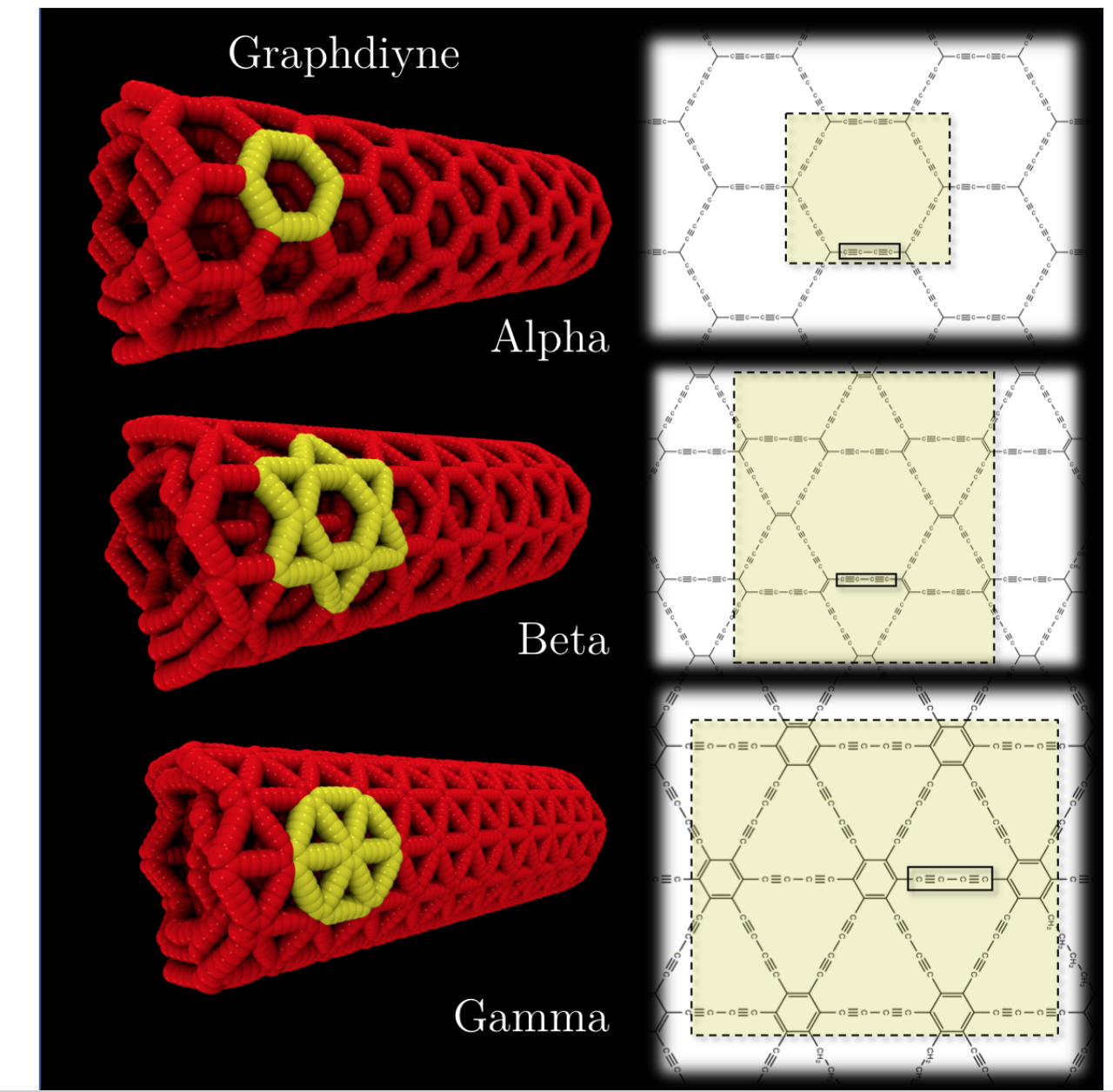
Solis, Daniel; Damasceno Borges, Daiane; Woellner, Cristiano; Galvao, Douglas
Structural and Thermal Stability of Graphyne and Graphdiyne Nanoscroll Structures (invited paper) Journal Article
In: ACS Applied Materials and Interfaces, vol. 11, pp. 2670−2676, 2019.
Abstract | Links | BibTeX | Tags: graphdiynes, Graphynes, Molecular Dynamics, Scrolls
@article{Solis2019,
title = {Structural and Thermal Stability of Graphyne and Graphdiyne Nanoscroll Structures (invited paper)},
author = {Solis, Daniel and Damasceno Borges, Daiane and Woellner, Cristiano and Galvao,
Douglas},
url = {https://pubs.acs.org/doi/10.1021/acsami.8b03481},
doi = {10.1021/acsami.8b03481},
year = {2019},
date = {2019-01-23},
journal = {ACS Applied Materials and Interfaces},
volume = {11},
pages = {2670−2676},
abstract = {Graphynes and graphdiynes are generic names for families of two-dimensional carbon allotropes, where acetylenic groups connect benzenoid-like hexagonal rings, with the coexistence of sp and sp2 hybridized carbon atoms. The main differences between graphynes and graphdiynes are the number of acetylenic groups (one and two for graphynes and graphdiynes, respectively). Similarly to graphene nanoscrolls, graphyne and graphdiynes nanoscrolls are nanosized membranes rolled into papyrus-like structures. In this work we studied through molecular dynamics simulations, using reactive potentials, the structural and thermal (up to 1000 K) stability of α,β,γ-graphyne and α,β,γ-graphdiyne scrolls. Our results demonstrate that stable nanoscrolls can be created for all the structures studied here, although they are less stable than corresponding graphene scrolls. This can be elucidated as a result of the higher graphyne/graphdiyne structural porosity in relation to graphene, and as a consequence, the π–π stacking interactions decrease.},
keywords = {graphdiynes, Graphynes, Molecular Dynamics, Scrolls},
pubstate = {published},
tppubtype = {article}
}
2018
Solis, Daniel; Borges, Daiane D.; Woellner, Cristiano F.; Galvao, Douglas S.
Structural and Thermal Stability of Graphyne and Graphdiyne Nanoscroll Structures Online
2018, visited: 02.03.2018, (preprint ArXiv: 1803.00154).
Abstract | Links | BibTeX | Tags: graphdiynes, Graphynes, molcular dynamics, Scrolls
@online{Solis2018b,
title = {Structural and Thermal Stability of Graphyne and Graphdiyne Nanoscroll Structures},
author = {Daniel Solis and Daiane D. Borges and Cristiano F. Woellner and Douglas S. Galvao},
url = {https://arxiv.org/abs/1803.00154},
year = {2018},
date = {2018-03-02},
urldate = {2018-03-02},
abstract = {Graphynes and graphdiynes are generic names for families of two-dimensional carbon allotropes,
where acetylenic groups connect benzenoid-like hexagonal rings, with the co-existence of sp and
sp
2 hybridized carbon atoms. The main differences between graphynes and graphdiynes are the
number of acetylenic groups (one and two for graphynes and graphdiynes, respectively).
Similarly to graphene nanoscrolls, graphyne and graphdiynes nanoscrolls are nanosized
membranes rolled up into papyrus-like structures. In this work we investigated through fully
atomistic reactive molecular dynamics simulations the structural and thermal (up to 1000K)
stability of α,β,γ-graphyne and α,β,γ-graphdiyne scrolls. Our results show that stable nanoscrolls
can be formed for all the structures investigated here, although they are less stable than
corresponding graphene scrolls. This can be explained as a consequence of the higher
graphyne/graphdiyne structural porosity in relation to graphene, which results in decreased π-π
stacking interactions. },
note = {preprint ArXiv: 1803.00154},
keywords = {graphdiynes, Graphynes, molcular dynamics, Scrolls},
pubstate = {published},
tppubtype = {online}
}
where acetylenic groups connect benzenoid-like hexagonal rings, with the co-existence of sp and
sp
2 hybridized carbon atoms. The main differences between graphynes and graphdiynes are the
number of acetylenic groups (one and two for graphynes and graphdiynes, respectively).
Similarly to graphene nanoscrolls, graphyne and graphdiynes nanoscrolls are nanosized
membranes rolled up into papyrus-like structures. In this work we investigated through fully
atomistic reactive molecular dynamics simulations the structural and thermal (up to 1000K)
stability of α,β,γ-graphyne and α,β,γ-graphdiyne scrolls. Our results show that stable nanoscrolls
can be formed for all the structures investigated here, although they are less stable than
corresponding graphene scrolls. This can be explained as a consequence of the higher
graphyne/graphdiyne structural porosity in relation to graphene, which results in decreased π-π
stacking interactions.
2016
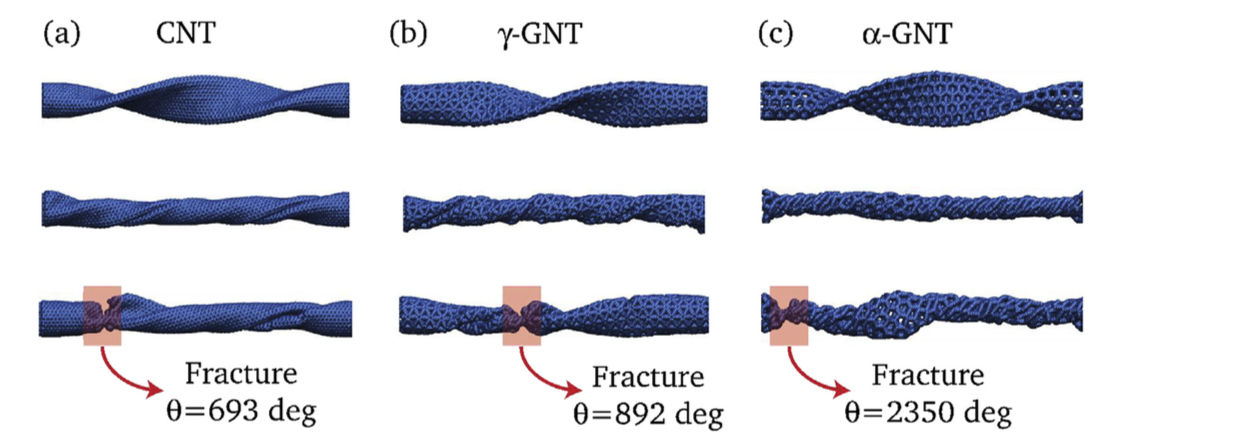
G. Brunetto J.M. de Sousa, V. R. Coluci
Torsional “superplasticity” of graphyne nanotubes Journal Article
In: Carbon, vol. 96, pp. 14-19, 2016.
Abstract | Links | BibTeX | Tags: Fracture, Graphynes, Mechanical Properties, Nanotubes
@article{deSousa2016,
title = {Torsional “superplasticity” of graphyne nanotubes},
author = {J.M. de Sousa, G. Brunetto, V.R. Coluci, D.S. Galvao },
url = {http://www.sciencedirect.com/science/article/pii/S000862231530258X},
doi = { http://dx.doi.org/10.1016/j.carbon.2015.09.039},
year = {2016},
date = {2016-01-01},
journal = {Carbon},
volume = {96},
pages = {14-19},
abstract = {Graphyne is a planar two-dimensional carbon allotrope formed by atoms in sp, sp2, and sp3 hybridized states. Topologically graphyne nanotubes (GNTs) can be considered as cylindrically rolled up graphyne sheets, similarly as carbon nanotubes (CNTs) can be considered rolled up graphene sheets. Due to the presence of single, double, and triple bonds, GNTs exhibit porous sidewalls that can be exploited in many diverse applications. In this work, we investigated the mechanical behavior of GNTs under torsional strains through reactive molecular dynamics simulations. Our results show that GNTs are more flexible than CNTs and exhibit “superplasticit”, with fracture angles that are up to 35 times higher than the ones reported to CNTs. This GNT “superplastic” behavior can be explained in terms of irreversible recon- struction processes (mainly associated with the triple bonds) that occur during torsional strains.},
keywords = {Fracture, Graphynes, Mechanical Properties, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
2015
Gustavo Brunetto Jose M. de Sousa, Vitor R. Coluci
Torsional "Superplasticity" of Graphyne Nanotubes Online
2015, (ArXiv reprint of Torsional "Superplasticity" of Graphyne Nanotubes, published in Carbon 96, 14 (2016).).
Abstract | Links | BibTeX | Tags: Allotropes, Graphynes, Mechanical Properties, Nanotubes
@online{deSousa2015b,
title = {Torsional "Superplasticity" of Graphyne Nanotubes},
author = {Jose M. de Sousa, Gustavo Brunetto, Vitor R. Coluci, Douglas S. Galvao},
url = {http://arxiv.org/abs/1509.08746},
year = {2015},
date = {2015-09-29},
abstract = {Graphyne is a planar two-dimensional carbon allotrope formed by atoms in sp, sp2, and sp3 hybridized states. Topologically graphyne nanotubes (GNTs) can be considered as cylindrically rolled up graphyne sheets, similarly as carbon nanotubes (CNTs) can be considered rolled up graphene sheets. Due to the presence of single, double, and triple bonds, GNTs exhibit porous sidewalls that can be exploited in many diverse applications. In this work, we investigated the mechanical behavior of GNTs under torsional strains through reactive molecular dynamics simulations. Our results show that GNTs are more flexible than CNTs and exhibit 'superplasticity', with fracture angles that are up to 35 times higher than the ones reported to CNTs. This GNT 'superplastic' behavior can be explained in terms of irreversible reconstruction processes (mainly associated with the triple bonds) that occur during torsional strains.},
note = {ArXiv reprint of Torsional "Superplasticity" of Graphyne Nanotubes, published in Carbon 96, 14 (2016).},
keywords = {Allotropes, Graphynes, Mechanical Properties, Nanotubes},
pubstate = {published},
tppubtype = {online}
}
Pedro A. S. Autreto, Douglas S. Galvao
Site dependent hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation Online
2015, (ArXiv draft of MRS Proceedings, 1726, mrsf14-1726-j02-02 (2015)).
Abstract | Links | BibTeX | Tags: Graphynes, Hydrogenation, Molecular Dynamics
@online{Autreto2015,
title = {Site dependent hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Pedro A. S. Autreto, Douglas S. Galvao},
url = {http://arxiv.org/abs/1501.04521},
year = {2015},
date = {2015-01-19},
journal = {arXiv preprint 1501.04521},
abstract = {Graphyne is a generic name for a carbon allotrope family of 2D structures, where acetylenic groups connect benzenoid rings, with the coexistence of sp and sp2 hybridized carbon atoms. In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics and structural changes of the hydrogenation of ALPHA, BETA, GAMMA graphyne forms. Our results showed that the existence of different sites for hydrogen bonding, related to single and triple bonds, makes the process of incorporating hydrogen atoms into graphyne membranes much more complex than the graphene ones. Our results also show that hydrogenation reactions are strongly site dependent and that the sp-hybridized carbon atoms are the preferential sites to chemical attacks. In our cases, the effectiveness of the hydrogenation (estimated from the number of hydrogen atoms covalently bonded to carbon atoms) follows the ALPHA, BETA, GAMMA graphyne structure ordering.},
note = {ArXiv draft of MRS Proceedings, 1726, mrsf14-1726-j02-02 (2015)},
keywords = {Graphynes, Hydrogenation, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}
Pedro A. S. Autreto, Douglas S. Galvao
Site Dependent Hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation Proceedings
vol. 1726, no. mrsf14-1726-j02-02, 2015, (MRS Proceedings, 1726, mrsf14-1726-j02-02 ).
Abstract | Links | BibTeX | Tags: Graphynes, Hydrogenation, Molecular Dynamics
@proceedings{Autreto2015b,
title = {Site Dependent Hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Pedro A. S. Autreto, Douglas S. Galvao},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=9702693&fulltextType=RA&fileId=S1946427415004649},
doi = {10.1557/opl.2015.464},
year = {2015},
date = {2015-01-01},
journal = {Mater. Res. Soc. Symp. Proc. },
volume = {1726},
number = {mrsf14-1726-j02-02},
abstract = {Graphyne is a generic name for a carbon allotrope family of 2D structures, where acetylenic groups connect benzenoid rings, with the coexistence of sp and sp2 hybridized carbon atoms. In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics and structural changes of the hydrogenation of α, β, and γ graphyne forms. Our results showed that the existence of different sites for hydrogen bonding, related to single and triple bonds, makes the process of incorporating hydrogen atoms into graphyne membranes much more complex than the graphene ones. Our results also show that hydrogenation reactions are strongly site dependent and that the sp-hybridized carbon atoms are the preferential sites to chemical attacks. In our cases, the effectiveness of the hydrogenation (estimated from the number of hydrogen atoms covalently bonded to carbon atoms) follows the α, β, γ-graphyne structure ordering.},
note = {MRS Proceedings, 1726, mrsf14-1726-j02-02 },
keywords = {Graphynes, Hydrogenation, Molecular Dynamics},
pubstate = {published},
tppubtype = {proceedings}
}
2014
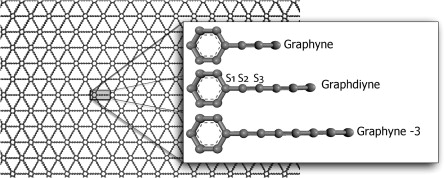
Autreto, PAS; de Sousa, JM; Galvao, DS
Site-dependent hydrogenation on graphdiyne Journal Article
In: Carbon, vol. 77, pp. 829–834, 2014.
Abstract | Links | BibTeX | Tags: Functionalization, Graphdyine, Graphene, Graphynes
@article{autreto2014site,
title = {Site-dependent hydrogenation on graphdiyne},
author = {Autreto, PAS and de Sousa, JM and Galvao, DS},
url = {http://www.sciencedirect.com/science/article/pii/S0008622314005429},
year = {2014},
date = {2014-01-01},
journal = {Carbon},
volume = {77},
pages = {829--834},
publisher = {Pergamon},
abstract = {Graphene is one of the most important materials in science today due to its unique and remarkable electronic, thermal and mechanical properties. However in its pristine state, graphene is a gapless semiconductor, what limits its use in transistor electronics. In part due to the revolution created by graphene in materials science, there is a renewed interest in other possible graphene-like two-dimensional structures. Examples of these structures are graphynes and graphdiynes, which are two-dimensional structures, composed of carbon atoms in sp2 and sp-hybridized states. Graphdiynes (benzenoid rings connecting two acetylenic groups) were recently synthesized and some of them are intrinsically nonzero gap systems. These systems can be easily hydrogenated and the relative level of hydrogenation can be used to tune the band gap values. We have investigated, using fully reactive molecular dynamics (ReaxFF), the structural and dynamics aspects of the hydrogenation mechanisms of graphdiyne membranes. Our results showed that the hydrogen bindings have different atom incorporation rates and that the hydrogenation patterns change in time in a very complex way. The formation of correlated domains reported to hydrogenated graphene is no longer observed in graphdiyne cases.},
keywords = {Functionalization, Graphdyine, Graphene, Graphynes},
pubstate = {published},
tppubtype = {article}
}
2013
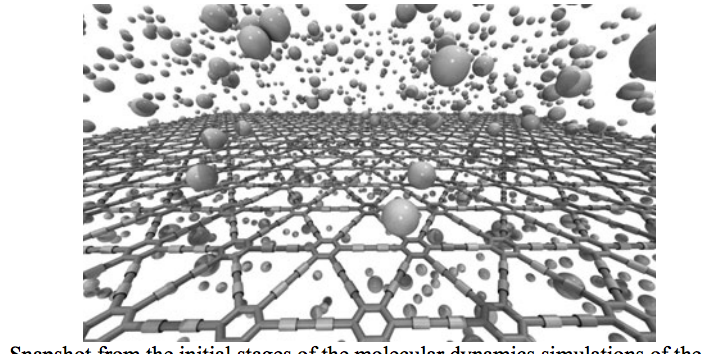
Autreto, PA; de Sousa, JM; Galvao, DS
On the Dynamics of Graphdiyne Hydrogenation Proceedings
Cambridge University Press, vol. 1549, 2013.
Abstract | Links | BibTeX | Tags: Graphdyine, Graphynes, Hydrogenation, Molecular Dynamics
@proceedings{autreto2013dynamics,
title = {On the Dynamics of Graphdiyne Hydrogenation},
author = {Autreto, PA and de Sousa, JM and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8915680&fileId=S1946427413006088},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1549},
pages = {59--64},
publisher = {Cambridge University Press},
abstract = {Graphene is a two-dimensional (2D) hexagonal array of carbon atoms in sp2-hybridized states. Graphene presents unique and exceptional electronic, thermal and mechanical properties. However, in its pristine state graphene is a gapless semiconductor, which poses some limitations to its use in some transistor electronics. Because of this there is a renewed interest in other possible two-dimensional carbon-based structures similar to graphene. Examples of this are graphynes and graphdiynes, which are two-dimensional structures, composed of carbon atoms in sp2 and sp-hybridized states. Graphdiynes (benzenoid rings connecting two acetylenic groups) were recently synthesized and they can be intrinsically nonzero gap systems. These systems can be easily hydrogenated and the amount of hydrogenation can be used to tune the band gap value. In this work we have investigated, through fully atomistic molecular dynamics simulations with reactive force field (ReaxFF), the structural and dynamics aspects of the hydrogenation mechanisms of graphdiyne membranes. Our results showed that depending on whether the atoms are in the benzenoid rings or as part of the acetylenic groups, the rates of hydrogenation are quite distinct and change in time in a very complex pattern. Initially, the most probable sites to be hydrogenated are the carbon atoms forming the triple bonds, as expected. But as the amount of hydrogenation increases in time this changes and then the carbon atoms forming single bonds become the preferential sites. The formation of correlated domains observed in hydrogenated graphene is no longer observed in the case of graphdiynes. We have also carried out ab initio DFT calculations for model structures in order to test the reliability of ReaxFF calculations.},
keywords = {Graphdyine, Graphynes, Hydrogenation, Molecular Dynamics},
pubstate = {published},
tppubtype = {proceedings}
}
2004
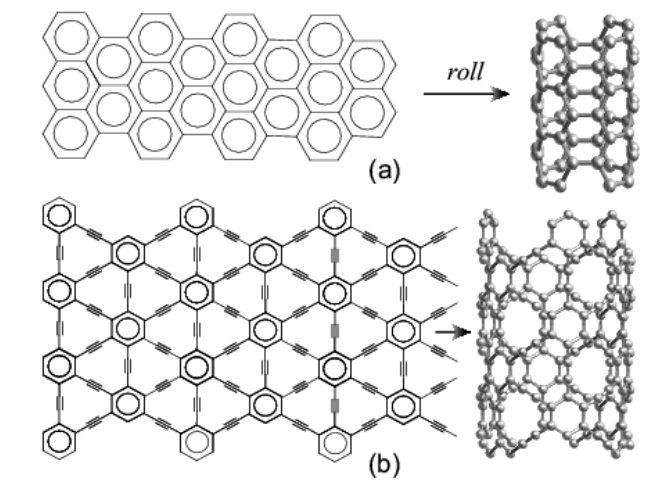
Coluci, VR; Braga, SF; Legoas, SB; Galvao, DS; Baughman, RH
New families of carbon nanotubes based on graphyne motifs Journal Article
In: Nanotechnology, vol. 15, no. 4, pp. S142, 2004.
Abstract | Links | BibTeX | Tags: Allotropes, Electronic Structure, Graphynes, Nanotubes
@article{coluci2004new,
title = {New families of carbon nanotubes based on graphyne motifs},
author = {Coluci, VR and Braga, SF and Legoas, SB and Galvao, DS and Baughman, RH},
url = {http://iopscience.iop.org/0957-4484/15/4/006},
year = {2004},
date = {2004-01-01},
journal = {Nanotechnology},
volume = {15},
number = {4},
pages = {S142},
publisher = {IOP Publishing},
abstract = {Electronic properties of proposed new families of carbon single walled nanotubes are investigated. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Analogous to ordinary nanotubes, armchair, zigzag and chiral graphyne nanotubes are possible. Tight-binding and ab initio density functional methods were used to predict the electronic properties of these unusual nanotubes. Of the three graphyne nanotube families analysed here, two provide metallic behaviour for armchair tubes and either metallic or semiconducting behaviour for zigzag nanotubes. For the other graphyne nanotube family investigated a diameter and chirality independent bandgap is predicted and a bandgap modulation study by structural distortions has been carried out for small longitudinal tube deformations. Interestingly, while the bandgap is insensitive to structure, the stress-induced bandgap changes can strongly depend both on the nanotube type and whether the strain is tensile or compressive.
},
keywords = {Allotropes, Electronic Structure, Graphynes, Nanotubes},
pubstate = {published},
tppubtype = {article}
}

Coluci, VR; Galvao, DS; Baughman, RH
Theoretical investigation of electromechanical effects for graphyne carbon nanotubes Journal Article
In: The Journal of chemical physics, vol. 121, no. 7, pp. 3228–3237, 2004.
Abstract | Links | BibTeX | Tags: Allotropes, Electroactuation, Electronic Structure, Graphynes, Nanotubes
@article{coluci2004theoretical,
title = {Theoretical investigation of electromechanical effects for graphyne carbon nanotubes},
author = {Coluci, VR and Galvao, DS and Baughman, RH},
url = {http://scitation.aip.org/content/aip/journal/jcp/121/7/10.1063/1.1772756},
year = {2004},
date = {2004-01-01},
journal = {The Journal of chemical physics},
volume = {121},
number = {7},
pages = {3228--3237},
publisher = {AIP Publishing},
abstract = {We present a theoretical study of the electronic and mechanical properties of graphyne-based nanotubes (GNTs). These semiconducting nanotubes result from the elongation of one-third of the covalent interconnections of graphite-based nanotubes by the introduction of yne groups. The effect of charge injection on the dimensions of GNTs was investigated using tight-binding calculations. Low amounts of electron injection are predicted to cause qualitatively different responses for armchair and zigzag graphyne nanotubes. Although the behavior is qualitatively similar to the usual carbon nanotubes, the charge-induced strains are predicted to be smaller for the GNTs than for ordinary single walled carbon nanotubes.},
keywords = {Allotropes, Electroactuation, Electronic Structure, Graphynes, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
2003
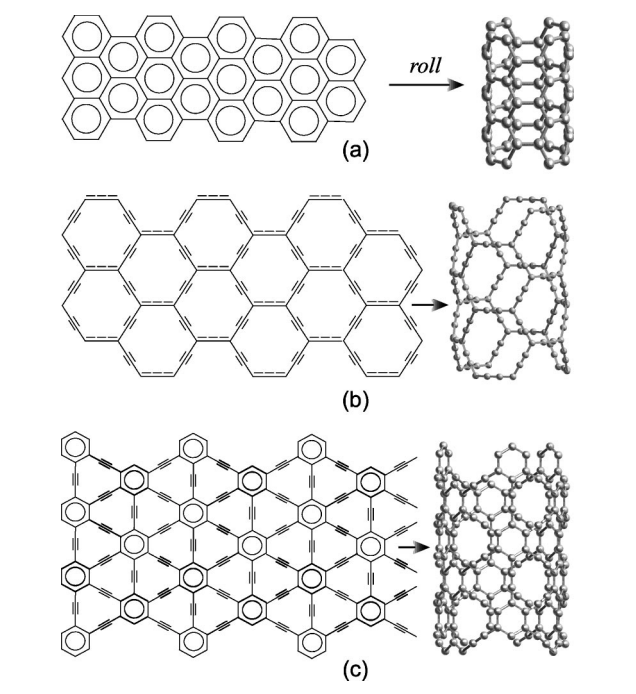
Coluci, VR; Braga, SF; Legoas, SB; Galvao, DS; Baughman, RH
Families of carbon nanotubes: Graphyne-based nanotubes Journal Article
In: Physical Review B, vol. 68, no. 3, pp. 035430, 2003.
Abstract | Links | BibTeX | Tags: Allotropes, Electronic Structure, Graphynes, Nanotubes
@article{coluci2003families,
title = {Families of carbon nanotubes: Graphyne-based nanotubes},
author = {Coluci, VR and Braga, SF and Legoas, SB and Galvao, DS and Baughman, RH},
url = {http://journals.aps.org/prb/abstract/10.1103/PhysRevB.68.035430},
year = {2003},
date = {2003-01-01},
journal = {Physical Review B},
volume = {68},
number = {3},
pages = {035430},
publisher = {APS},
abstract = {New families of carbon single-walled nanotubes are proposed and their electronic structures are investigated. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Analogously to ordinary nanotubes, armchair, zigzag, and chiral graphyne nanotubes are possible. We here predict the electronic properties of these unusual nanotubes using tight-binding and ab initio density functional methods. Of the three graphyne nanotube families analyzed here, two provide metallic behavior for armchair tubes and either metallic or semiconducting behavior for zigzag nanotubes. A diameter- and chirality-independent band gap is predicted for the other investigated graphyne family, as well as an oscillatory dependence of the effective mass on nanotube diameter.},
keywords = {Allotropes, Electronic Structure, Graphynes, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
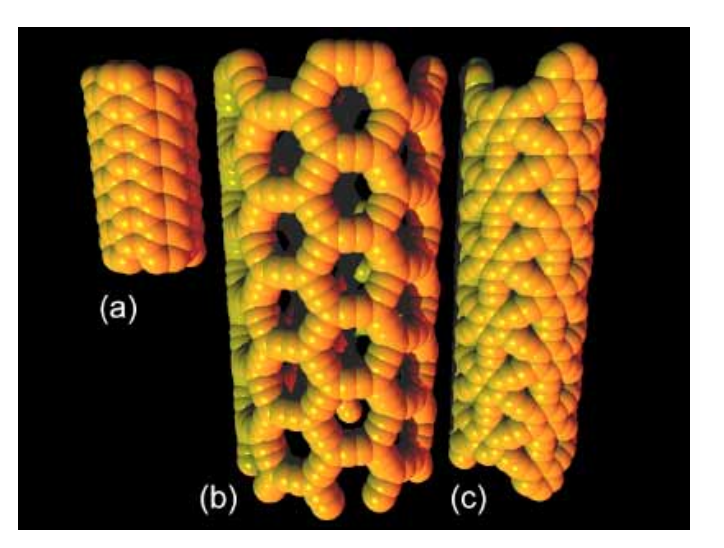
Coluci, VR; Braga, SF; Legoas, SB; Galvao, DS; Baughman, RH
Graphyne Nanotubes: New Families of Carbon Nanotubes Proceedings
Warrendale, Pa.; Materials Research Society; 1999, vol. 739, 2003.
Abstract | Links | BibTeX | Tags: Allotropes, Electronic Structure, Graphynes, Nanotubes
@proceedings{coluci2003graphyne,
title = {Graphyne Nanotubes: New Families of Carbon Nanotubes},
author = {Coluci, VR and Braga, SF and Legoas, SB and Galvao, DS and Baughman, RH},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8031794},
year = {2003},
date = {2003-01-01},
journal = {MATERIALS RESEARCH SOCIETY SYMPOSIUM PROCEEDINGS},
volume = {739},
pages = {175--180},
publisher = {Warrendale, Pa.; Materials Research Society; 1999},
abstract = {Fundamentally new families of carbon single walled nanotubes are proposed. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Similarly to ordinary nanotubes, armchair, zig-zag, and chiral graphyne nanotubes are possible. We present here results for the electronic properties of graphyne based tubes obtained from tight-binding and ab initio density functional methods.},
keywords = {Allotropes, Electronic Structure, Graphynes, Nanotubes},
pubstate = {published},
tppubtype = {proceedings}
}
2002
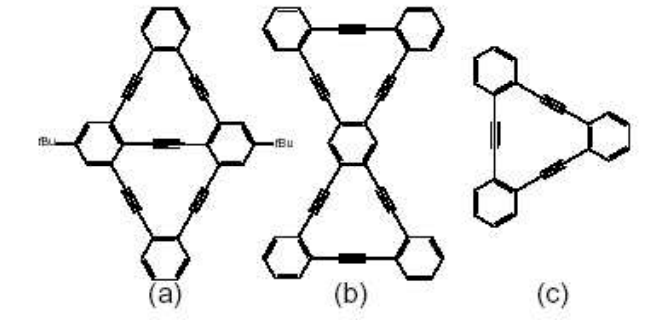
Coluci, V R; Braga, SF; Legoas, Sergio B; Galvao, Douglas S; Baughman, RH
New families of carbon nanotubes Journal Article
In: arXiv preprint cond-mat/0207085, 2002.
Abstract | Links | BibTeX | Tags: Allotropes, Electronic Structure, Graphynes, Nanotubes
@article{coluci2002new,
title = {New families of carbon nanotubes},
author = {Coluci, V R and Braga, SF and Legoas, Sergio B and Galvao, Douglas S and Baughman, RH},
url = {http://arxiv.org/abs/cond-mat/0207085},
year = {2002},
date = {2002-01-01},
journal = {arXiv preprint cond-mat/0207085},
abstract = {Fundamentally new families of carbon single walled nanotubes are proposed. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Similarly to ordinary nanotubes, arm-chair, zig-zag, and chiral graphyne nanotubes are possible. Electronic properties, predicted using tight-binding and ab initio density functional methods, show a rich variety of metallic and semiconducting behaviors.},
keywords = {Allotropes, Electronic Structure, Graphynes, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ

