
Malviya, Kirtman D; Oliveira, Eliezer F; Autreto, Pedro A S; Ajayan, Pulickel M; Galvao, D S; Tiwary, Candra S; Chattopadhyay, Kumanio
Mixing the immiscible through high-velocity mechanical impacts: an experimental and theoretical study Journal Article
In: Journal of Physics D: Applied Physics, vol. 52, no. 44, pp. 445304, 2019.
@article{Malviya2019,
title = {Mixing the immiscible through high-velocity mechanical impacts: an experimental and theoretical study},
author = {Malviya, Kirtman D and Oliveira, Eliezer F and Autreto, Pedro A S and Ajayan, Pulickel M and Galvao, D S and Tiwary, Candra S and Chattopadhyay, Kumanio},
url = {https://iopscience.iop.org/article/10.1088/1361-6463/ab36d1/meta},
doi = {10.1088/1361-6463/ab36d1},
year = {2019},
date = {2019-08-20},
journal = {Journal of Physics D: Applied Physics},
volume = {52},
number = {44},
pages = {445304},
abstract = {In two-component metallic systems, thermodynamic immiscibility leads to phase separation
such as in two-phase eutectic compositional alloys. The limit of the immiscibility of
component elements under non-equilibrium conditions have been explored, but achieving
complete miscibility and formation of single phase microstructures in eutectic alloys would
be unprecedented. Here we report that during low-temperature ball milling that provides high
energy impact, complete mixing of phases can occur in immiscible Ag-Cu eutectic alloys.
From combined theoretical and experimental studies, we show that impact can produce solid
solutions of Ag-Cu nanoparticles of eutectic composition. Our results show that phase
diagrams of low dimensional materials under non-equilibrium conditions remain unexplored
and could lead to new alloy microstructures drastically different from their bulk counterparts.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
such as in two-phase eutectic compositional alloys. The limit of the immiscibility of
component elements under non-equilibrium conditions have been explored, but achieving
complete miscibility and formation of single phase microstructures in eutectic alloys would
be unprecedented. Here we report that during low-temperature ball milling that provides high
energy impact, complete mixing of phases can occur in immiscible Ag-Cu eutectic alloys.
From combined theoretical and experimental studies, we show that impact can produce solid
solutions of Ag-Cu nanoparticles of eutectic composition. Our results show that phase
diagrams of low dimensional materials under non-equilibrium conditions remain unexplored
and could lead to new alloy microstructures drastically different from their bulk counterparts.
de Sousa, Jose Moreira; Autreto, Pedro da Silva; Galvao, Douglas Soares
Hydrogenation Dynamics Process of Single-wall Carbon Nanotube Twisted (under review) Journal Article
In: 2019.
@article{deSousa2019d,
title = {Hydrogenation Dynamics Process of Single-wall Carbon Nanotube Twisted (under review)},
author = {de Sousa, Jose Moreira and Autreto, Pedro da Silva and Galvao, Douglas Soares},
year = {2019},
date = {2019-07-15},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
JM; Sousa, Bizao
Elastic Properties of Graphyne-based Nanotubes Online
2019, (ArXiv preprint.).
@online{deSousa2019b,
title = {Elastic Properties of Graphyne-based Nanotubes},
author = {de Sousa, JM; , Bizao, RA; Sousa Filho, VP; Aguiar, AL; Coluci, VR; Pugno, NM; Girao, EC; Souza Filho, AG; Galvao, DS},
url = {https://arxiv.org/pdf/1905.02104.pdf},
year = {2019},
date = {2019-04-07},
abstract = {Graphyne nanotubes (GNTs) are nanostructures obtained from rolled up graphyne sheets,
in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes
are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to
conventional CNTs, GNTs can present different chiralities and electronic properties. Because
of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their
mechanical properties. In this work, we studied the mechanical response of GNTs under
tensile stress using fully atomistic molecular dynamics simulations and density functional
theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs
at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller
ultimate strength and Young’s modulus values. This is a consequence of the combined
effects of the existence of triple bonds and increased porosity/flexibility due to the presence
of acetylenic groups.},
note = {ArXiv preprint.},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes
are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to
conventional CNTs, GNTs can present different chiralities and electronic properties. Because
of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their
mechanical properties. In this work, we studied the mechanical response of GNTs under
tensile stress using fully atomistic molecular dynamics simulations and density functional
theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs
at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller
ultimate strength and Young’s modulus values. This is a consequence of the combined
effects of the existence of triple bonds and increased porosity/flexibility due to the presence
of acetylenic groups.
JM; Sousa, Bizao
Elastic Properties of Graphyne-Based Nanotubes Journal Article
In: Computational Materials Science, vol. 170, pp. 109153, 2019.
@article{deSousa2019c,
title = {Elastic Properties of Graphyne-Based Nanotubes},
author = {de Sousa, JM; , Bizao, RA; Sousa Filho, VP; Aguiar, AL; Coluci, VR; Pugno, NM; Girao, EC; Souza Filho, AG; Galvao, DS},
url = {https://www.sciencedirect.com/science/article/pii/S0927025619304525?dgcid=coauthor#s0040},
doi = {10.1016/j.commatsci.2019.109153},
year = {2019},
date = {2019-04-03},
journal = {Computational Materials Science},
volume = {170},
pages = {109153},
abstract = {Graphyne nanotubes (GNTs) are nanostructures obtained from rolled up graphyne sheets, in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to conventional CNTs, GNTs can present different chiralities and electronic properties. Because of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their mechanical properties. In this work, we studied the mechanical response of GNTs under tensile stress using fully atomistic molecular dynamics simulations and density functional theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller ultimate strength and Young’s modulus values. This is a consequence of the combined effects of the existence of triple bonds and increased porosity/flexibility due to the presence of acetylenic groups.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Fonseca, AF; Dantas, SO; Galvao, DS; Zhang, D; Sinnott, SB
The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review) Journal Article
In: 2019.
@article{Fonseca2019d,
title = {The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review)},
author = {Fonseca, AF and Dantas, SO and Galvao, DS and Zhang, D and Sinnott, SB},
year = {2019},
date = {2019-04-03},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Arpan; Gumaste Rout, Anurag; Pandey
Bio-inspired Aluminum Composite reinforced with Soft polymer with enhanced strength and plasticity (under review) Journal Article
In: 2019.
@article{Rout2019,
title = {Bio-inspired Aluminum Composite reinforced with Soft polymer with enhanced strength and plasticity (under review)},
author = {Rout, Arpan; Gumaste, Anurag; Pandey, Praful; Oliveira, Eliezer; Demiss,
Solomon; P., Mahesh; Bhatt, Chintan; Raphael, Kiran; Ayyagari, Ravi; Autreto, Pedro;
Palit, Mithun; Femi, Olu Emmanuel; Galvao, Douglas; Arora, Amit; Tiwary, Chandra},
year = {2019},
date = {2019-03-30},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
AF; Dantas Fonseca, SO; Galvao
The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study Online
2019, (ArXiv preprint).
@online{Fonseca2019b,
title = {The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study},
author = {Fonseca, AF; Dantas, SO; Galvao, DS; Zhang, D; Sinnott SB},
url = {https://arxiv.org/pdf/1904.09871.pdf},
year = {2019},
date = {2019-03-22},
abstract = {Two experimental studies reported the spontaneous formation of amorphous and crystalline
structures of C60 intercalated between graphene and a substrate. They observed interesting
phenomena ranging from reaction between C60 molecules under graphene to graphene
sagging between the molecules and control of strain in graphene. Motivated by these works,
we performed fully atomistic reactive molecular dynamics simulations to study the formation
and thermal stability of graphene wrinkles as well as graphene attachment to and detachment
from the substrate when graphene is laid over a previously distributed array of C60 molecules
on a copper substrate at different values of temperature. As graphene compresses the C60
molecules against the substrate, and graphene attachment to the substrate between C60s
(“C60S” stands for plural of C60) depends on the height of graphene wrinkles, configurations
with both frozen and non-frozen C60s structures were investigated in order to verify the
experimental result of stable sagged graphene when the distance between C60s is about 4 nm
and height of graphene wrinkles is about 0.8 nm. Below the distance of 4 nm between C60s,
graphene becomes locally suspended and less strained. We show that this happens when C60s
are allowed to deform under the compressive action of graphene. If we keep the C60s frozen,
spontaneous “blanketing” of graphene happens only when the distance between them are
equal or above 7 nm. Both above results for the existence of stable sagged graphene for C60
distances of 4 or 7 nm are shown to agree with a mechanical model relating the rigidity of
graphene to the energy of graphene-substrate adhesion. Although the studies of intercalation
of molecules on interfaces formed by graphene-substrate are motivated by finding out ways to
control wrinkling and strain in graphene, our work reveals the shape and structure of
intercalated molecules and the role of stability and wrinkling on final structure of graphene.
In particular, this study might help the development of 2D confined nanoreactors that are
considered in literature to be the next advanced step on chemical reactions.},
note = {ArXiv preprint},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
structures of C60 intercalated between graphene and a substrate. They observed interesting
phenomena ranging from reaction between C60 molecules under graphene to graphene
sagging between the molecules and control of strain in graphene. Motivated by these works,
we performed fully atomistic reactive molecular dynamics simulations to study the formation
and thermal stability of graphene wrinkles as well as graphene attachment to and detachment
from the substrate when graphene is laid over a previously distributed array of C60 molecules
on a copper substrate at different values of temperature. As graphene compresses the C60
molecules against the substrate, and graphene attachment to the substrate between C60s
(“C60S” stands for plural of C60) depends on the height of graphene wrinkles, configurations
with both frozen and non-frozen C60s structures were investigated in order to verify the
experimental result of stable sagged graphene when the distance between C60s is about 4 nm
and height of graphene wrinkles is about 0.8 nm. Below the distance of 4 nm between C60s,
graphene becomes locally suspended and less strained. We show that this happens when C60s
are allowed to deform under the compressive action of graphene. If we keep the C60s frozen,
spontaneous “blanketing” of graphene happens only when the distance between them are
equal or above 7 nm. Both above results for the existence of stable sagged graphene for C60
distances of 4 or 7 nm are shown to agree with a mechanical model relating the rigidity of
graphene to the energy of graphene-substrate adhesion. Although the studies of intercalation
of molecules on interfaces formed by graphene-substrate are motivated by finding out ways to
control wrinkling and strain in graphene, our work reveals the shape and structure of
intercalated molecules and the role of stability and wrinkling on final structure of graphene.
In particular, this study might help the development of 2D confined nanoreactors that are
considered in literature to be the next advanced step on chemical reactions.
AF; Dantas Fonseca, SO; Galvao
The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review) Journal Article
In: 2019.
@article{Fonseca2019c,
title = {The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review)},
author = {Fonseca, AF; Dantas, SO; Galvao, DS; Zhang, D; Sinnott SB},
year = {2019},
date = {2019-03-15},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Routa, Arpan; Pandeyb, Praful; Oliveira, Eliezer Fernando; da Silva Autreto, Pedro Alves; Gumastea, Anurag; Singha, Amit; Galvao, Douglas Soares; Aroraa, Amit; Tiwary, Chandra Sekhar
Atomically locked interfaces of metal (Aluminum) and Polymer (Polypropylene) using mechanical friction Journal Article
In: Polymer, vol. 169, pp. 148-153, 2019.
@article{Routa2019,
title = {Atomically locked interfaces of metal (Aluminum) and Polymer (Polypropylene) using mechanical friction},
author = {Arpan Routa and Praful Pandeyb and Eliezer Fernando Oliveira and Pedro Alves da Silva Autreto and Anurag Gumastea and Amit Singha and Douglas Soares Galvao and Amit Aroraa and Chandra Sekhar Tiwary},
year = {2019},
date = {2019-02-23},
journal = {Polymer},
volume = {169},
pages = {148-153},
abstract = {Joining different parts is one of a crucial component of designing/engineering of materials. The current energy, low efficiency weight automotive and aerospace components commonly consist of different class of materials, such as metal, polymer, and ceramics, etc. Joining these components remains a challenge. Here, we demonstrate joining of metal (aluminum) and polymer (PP) using mechanical friction. The detailed characterisation demonstrates that atomically locked interfaces are formed in such joining without the presence of any chemical bond at the interfaces. The waterproof and strong interface is formed in such process. Fully atomistic molecular dynamics simulations were also carried out to provide further insights on these mechanisms.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Eliezer F; Autreto Oliveira, Pedro AS; Woellner
On the mechanical properties of protomene: A theoretical investigation Journal Article
In: Computational Materials Science, vol. 161, pp. 190-198, 2019.
@article{Oliveira2019c,
title = {On the mechanical properties of protomene: A theoretical investigation},
author = {Oliveira, Eliezer F; Autreto, Pedro AS; Woellner, Cristiano F; Galvao, Douglas S},
year = {2019},
date = {2019-02-07},
journal = {Computational Materials Science},
volume = {161},
pages = {190-198},
abstract = {We report a detailed study through fully atomistic molecular dynamics simulations and DFT calculations on the mechanical properties of protomene. Protomene is a new carbon allotrope composed of a mixture of sp2 and sp3 hybridized states. Our results indicate that protomene presents an anisotropic behavior about tensile deformations. At room temperature, protomene presents an ultimate strength of ~100 GPa and Young's modulus of ~600 GPa, lower than the same for other carbon allotropes. Despite that, protomente presents the highest ultimate strain along the z-direction (~ 24.7%). Our results also show that stretching the protomene along the z-direction or heating it can induce a semiconductor-metallic phase transition, due to a high amount of sp3 bonds that are converted to sp2 ones.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Sanjit; Ozden Bhowmick, Sehmus; Bizão
High temperature quasistatic and dynamic mechanical behavior of interconnected 3D carbon nanotube structures Journal Article
In: Carbon, vol. 142, pp. 291-299, 2019.
@article{Bhowmick2019,
title = {High temperature quasistatic and dynamic mechanical behavior of interconnected 3D carbon nanotube structures},
author = {Bhowmick, Sanjit; Ozden, Sehmus; Bizão, Rafael A; Machado, Leonardo Dantas; Asif, SA Syed; Pugno, Nicola M; Galvao, Douglas S; Tiwary, Chandra Sekhar; Ajayan, PM},
url = {https://www.sciencedirect.com/science/article/pii/S0008622318308911},
doi = {10.1016/j.carbon.2018.09.075},
year = {2019},
date = {2019-02-01},
journal = {Carbon},
volume = {142},
pages = {291-299},
abstract = {Carbon nanotubes (CNTs) are one of the most appealing materials in recent history for both research and commercial interest because of their outstanding physical, chemical, and electrical properties. This is particularly true for 3D arrangements of CNTs which enable their use in larger scale devices and structures. In this paper, the effect of temperature on the quasistatic and dynamic deformation behavior of 3D CNT structures is presented for the first time. An in situ high-temperature nanomechanical instrument was used inside an SEM at high vacuum to investigate mechanical properties of covalently interconnected CNT porous structures in a wide range of temperature. An irreversible bucking at the base of pillar samples was found as a major mode of deformation at room and elevated temperatures. It has been observed that elastic modulus and critical load to first buckle formation decrease progressively with increasing temperature from 25 °C to 750 °C. To understand fatigue resistance, pillars made from this unique structure were compressed to 100 cycles at room temperature and 750 °C. While the structure showed remarkable resistance to fatigue at room temperature, high temperature significantly lowers fatigue resistance. Molecular dynamics (MD) simulation of compression highlights the critical role played by covalent interconnections which prevent localized bending and improve mechanical properties.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Solis, Daniel; Damasceno Borges, Daiane; Woellner, Cristiano; Galvao, Douglas
Structural and Thermal Stability of Graphyne and Graphdiyne Nanoscroll Structures (invited paper) Journal Article
In: ACS Applied Materials and Interfaces, vol. 11, pp. 2670−2676, 2019.
@article{Solis2019,
title = {Structural and Thermal Stability of Graphyne and Graphdiyne Nanoscroll Structures (invited paper)},
author = {Solis, Daniel and Damasceno Borges, Daiane and Woellner, Cristiano and Galvao,
Douglas},
url = {https://pubs.acs.org/doi/10.1021/acsami.8b03481},
doi = {10.1021/acsami.8b03481},
year = {2019},
date = {2019-01-23},
journal = {ACS Applied Materials and Interfaces},
volume = {11},
pages = {2670−2676},
abstract = {Graphynes and graphdiynes are generic names for families of two-dimensional carbon allotropes, where acetylenic groups connect benzenoid-like hexagonal rings, with the coexistence of sp and sp2 hybridized carbon atoms. The main differences between graphynes and graphdiynes are the number of acetylenic groups (one and two for graphynes and graphdiynes, respectively). Similarly to graphene nanoscrolls, graphyne and graphdiynes nanoscrolls are nanosized membranes rolled into papyrus-like structures. In this work we studied through molecular dynamics simulations, using reactive potentials, the structural and thermal (up to 1000 K) stability of α,β,γ-graphyne and α,β,γ-graphdiyne scrolls. Our results demonstrate that stable nanoscrolls can be created for all the structures studied here, although they are less stable than corresponding graphene scrolls. This can be elucidated as a result of the higher graphyne/graphdiyne structural porosity in relation to graphene, and as a consequence, the π–π stacking interactions decrease.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
de Sousa, JM; Aguiar, AL; Girao, EC; Fonseca, Alexandre F; AG Filho, Souza; Galvao, Douglas S
Mechanical Properties and Fracture Patterns of Pentagraphene Membranes (under review) Journal Article
In: 2019.
@article{deSousa2019,
title = {Mechanical Properties and Fracture Patterns of Pentagraphene Membranes (under review)},
author = {de Sousa, JM and Aguiar, AL and Girao, EC and Fonseca, Alexandre F and AG Filho, Souza and Galvao, Douglas S},
year = {2019},
date = {2019-01-05},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Fonseca, Alexandre F.; Galvao, Douglas S.
Self-tearing and self-peeling of folded graphene nanoribbons Journal Article
In: Carbon, vol. 143, pp. 230-239, 2019.
@article{Fonseca2019,
title = {Self-tearing and self-peeling of folded graphene nanoribbons},
author = {Alexandre F. Fonseca and Douglas S. Galvao},
url = {https://www.sciencedirect.com/science/article/pii/S0008622318310431},
doi = {10.1016/j.carbon.2018.11.020},
year = {2019},
date = {2019-01-05},
journal = {Carbon},
volume = {143},
pages = {230-239},
abstract = {A recent experimental study showed that an induced folded flap of graphene can spontaneously drive itself its tearing and peeling off a substrate, thus producing long, micrometer sized, regular trapezoidal-shaped folded graphene nanoribbons. As long as the size of the graphene flaps is above a threshold value, the “tug of war” between the forces of adhesion of graphene-graphene and graphene-substrate, flexural strain of folded region and carbon-carbon (C-C) covalent bonds favor the self-tearing and self-peeling off process. As the detailed information regarding the atomic scale mechanism involved in the process remains not fully understood, we carried out atomistic reactive molecular dynamics simulations to address some features of the process. We show that large thermal fluctuations can prevent the process by increasing the probability of chemical reactions between carbon dangling bonds of adjacent graphene layers. The effects of the strength of attraction between graphene and the substrate on the ribbon growth velocities at the early stages of the phenomenon were also investigated. Structures with initial armchair crack-edges were observed to form more uniform cuts than those having initial zigzag ones. Our results are of importance to help set up new experiments on this phenomenon, especially with samples with nanoscale sized cuts. },
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Sean P; Perim Collins, Eric; Daff
Idealized Carbon-Based Materials Exhibiting Record Deliverable Capacities for Vehicular Methane Storage Journal Article
In: The Journal of Physical Chemistry C, vol. 123, pp. 1050-1058, 2019.
@article{Collins2019,
title = {Idealized Carbon-Based Materials Exhibiting Record Deliverable Capacities for Vehicular Methane Storage},
author = {Collins, Sean P; Perim, Eric; Daff, Thomas D; Skaf, Munir S; Galvao, Douglas Soares; Woo, Tom K},
url = {https://pubs.acs.org/doi/abs/10.1021/acs.jpcc.8b09447},
doi = {10.1021/acs.jpcc.8b09447},
year = {2019},
date = {2019-01-05},
journal = {The Journal of Physical Chemistry C},
volume = {123},
pages = {1050-1058},
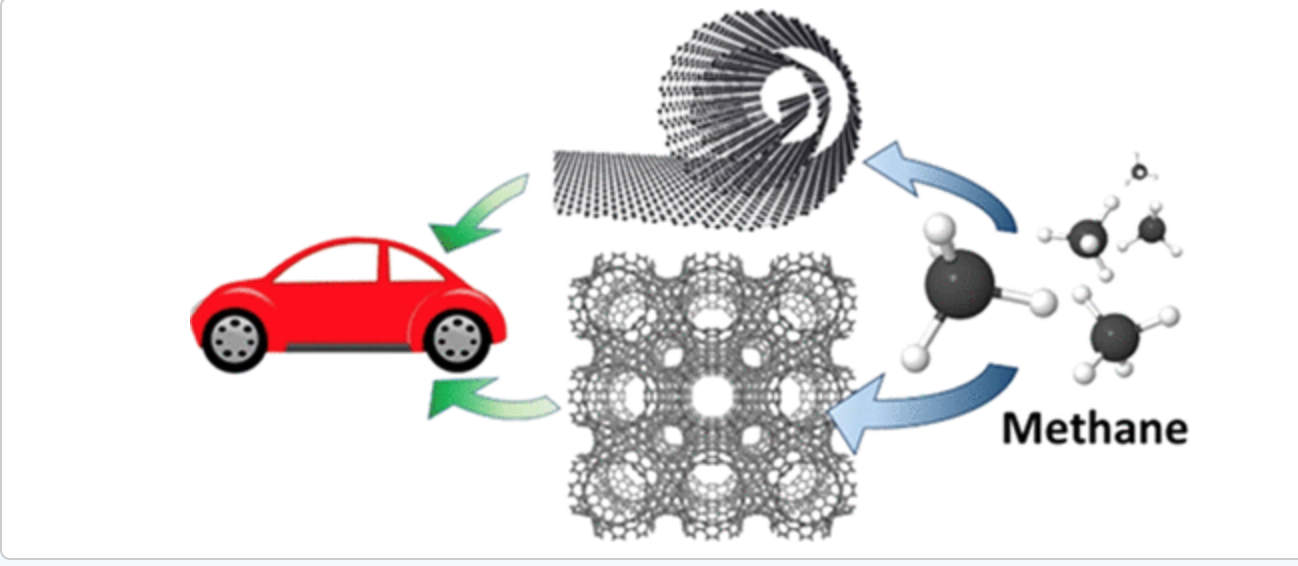
abstract = {Materials for vehicular methane storage have been extensively studied, although no suitable material has been found. In this work, we use molecular simulation to investigate three types of carbon-based materials, Schwarzites, layered graphenes, and carbon nanoscrolls, for use in vehicular methane storage under adsorption conditions of 65 bar and 298 K and desorption conditions of 5.8 bar and 358 K. Ten different Schwarzites were tested and found to have high adsorption with maximums at 273 VSTP/V, but middling deliverable capacities of no more than 131 VSTP/V. Layered graphene and graphene nanoscrolls were found to have extremely high CH4 adsorption capacities of 355 and 339 VSTP/V, respectively, when the interlayer distance was optimized to 11 Å. The deliverable capacities of perfectly layered graphene and graphene nanoscrolls were also found to be exceptional with values of 266 and 252 VSTP/V, respectively, with optimized interlayer distances. These values make idealized graphene and nanoscrolls the record holders for adsorption and deliverable capacities under vehicular methane storage conditions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Owuor, Peter Samora; Inthong, Suchittra; Sajadi, Seyed Mohammad; Intawin, Pratthana; Chipara, Alin C.; Woellner, Cristiano F.; Sayed, Farheen N.; Tsang, Harvey H.; Stender, Anthony; Vajtai, Robert; Pengpat, Kamonpan; Eitssayeam, Sukum; Galvao, Douglas S.; Lou, Jun; Tiwary, Chandra Sekhar; Ajayan, Pulickel M.
Elastic and ‘transparent bone’ as an electrochemical separator Journal Article
In: Materials Chemistry Today, vol. 12, pp. 132-138, 2019.
@article{Owuor2019,
title = {Elastic and ‘transparent bone’ as an electrochemical separator},
author = {Peter Samora Owuor and Suchittra Inthong and Seyed Mohammad Sajadi and Pratthana Intawin and Alin C. Chipara and Cristiano F. Woellner and Farheen N. Sayed and Harvey H. Tsang and Anthony Stender and Robert Vajtai and Kamonpan Pengpat and Sukum Eitssayeam and Douglas S. Galvao and Jun Lou and Chandra Sekhar Tiwary and Pulickel M. Ajayan},
url = {https://reader.elsevier.com/reader/sd/pii/S246851941830291X?token=B3C1F35B7DCEA8636EFB32B8D1D71EEC9852E58D0729A622DAFDF86C3EE65DF2A33E77CE7534A5D66D3854C396F69D1A},
doi = {10.1016/j.mtchem.2018.12.009},
year = {2019},
date = {2019-01-05},
journal = {Materials Chemistry Today},
volume = {12},
pages = {132-138},
abstract = {Organic matrix of bone mainly composed of collagen matrix serve as a crucial component for remarkable toughness and strength in bones. The porous collagen matrix can also serve as efficient template for various applications such as nanoparticles synthetic, catalysis or catalysis supports, electrochemical separator, filtration membrane and tissue engineering. However, fabricating collagen matrix from bones without degrading its morphological structure still remain a challenge. Here we present evidence of how ceramic crystals from a bone can be removed to fabricate a complete ‘transparent bone’ structure with improved porous and elasticity. We show that demineralization or selective etching using dilute acid (citric) can remove ceramics mineral nanoparticles without degrading the collagen matrix. The transparent bone collagen matrix is investigated as the separator in electrochemical supercapacitor with aqueous electrolyte where it shows better performance compared to conventional separators.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Eliezer F; Autreto Oliveira, Pedro AS; Woellner
Mechanical Properties of Protomene: A Molecular Dynamics Investigation Journal Article
In: MRS Advances, 2019.
@article{Oliveira2019,
title = {Mechanical Properties of Protomene: A Molecular Dynamics Investigation},
author = {Oliveira, Eliezer F; Autreto, Pedro AS; Woellner, Cristiano F; Galvao, Douglas S},
url = {www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-protomene-a-molecular-dynamics-investigation/CBAC89BDB5942E3353A5C00BD5D0D9CA},
doi = {10.1557/adv.2018.670},
year = {2019},
date = {2019-01-05},
journal = {MRS Advances},
abstract = {Recently, a new class of carbon allotrope called protomene was proposed. This new structure is composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3 carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now, its mechanical properties have not been investigated. In this work, we have investigated protomene mechanical behavior under tensile strain through fully atomistic reactive molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS code. At room temperature, our results show that the protomene is very stable and the obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical fracture.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nakar, Dekel; Gordeev, Georgy; Machado, Leonardo D.; Popovitz-Biro, Ronit; Rechav, Katya; Oliveira, Eliezer F.; Kusch, Patryk; Jorio, Ado; Galvao, Douglas S.; Reich, Stephanie; Joselevich, Ernesto
Few-Wall Carbon Nanotube Coils (under review) Journal Article
In: 2019.
@article{Nakar2019,
title = {Few-Wall Carbon Nanotube Coils (under review)},
author = {Dekel Nakar and Georgy Gordeev and Leonardo D. Machado and Ronit Popovitz-Biro and Katya Rechav and Eliezer F. Oliveira and Patryk Kusch and Ado Jorio and Douglas S. Galvao and Stephanie Reich and Ernesto Joselevich},
year = {2019},
date = {2019-01-01},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Pedro AS Autreto Eliezer F Oliveira, Cristiano F Woellner
Mechanical Properties of Protomene: A Molecular Dynamics Investigation Online
2018, (preprint arXiv:1810.09924v1 ).
@online{Oliveira2018g,
title = {Mechanical Properties of Protomene: A Molecular Dynamics Investigation},
author = {Eliezer F Oliveira, Pedro AS Autreto, Cristiano F Woellner, Douglas S Galvao},
url = {https://arxiv.org/abs/1810.09924},
year = {2018},
date = {2018-10-23},
abstract = {Recently, a new class of carbon allotrope called protomene was proposed. This new structure is
composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3
carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations
have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now,
its mechanical properties have not been investigated. In this work, we have investigated
protomene mechanical behavior under tensile strain through fully atomistic reactive
molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS
code. At room temperature, our results show that the protomene is very stable and the
obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest
ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate
strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical
fracture.},
note = {preprint arXiv:1810.09924v1 },
keywords = {},
pubstate = {published},
tppubtype = {online}
}
composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3
carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations
have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now,
its mechanical properties have not been investigated. In this work, we have investigated
protomene mechanical behavior under tensile strain through fully atomistic reactive
molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS
code. At room temperature, our results show that the protomene is very stable and the
obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest
ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate
strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical
fracture.
Alexandre F. Fonseca, Douglas S. Galvao
Self-tearing and self-peeling of folded graphene nanoribbons Online
2018, (preprint arXiv:1808.08872).
@online{Fonseca2018d,
title = { Self-tearing and self-peeling of folded graphene nanoribbons},
author = {Alexandre F. Fonseca, Douglas S. Galvao
},
url = {https://arxiv.org/abs/1808.08872},
year = {2018},
date = {2018-08-27},
abstract = {A recent experimental study showed that an induced folded flap of graphene can spontaneously drive itself its tearing and peeling off a substrate, thus producing long, micrometer sized, regular trapezoidal-shaped folded graphene nanoribbons. As long as the size of the graphene flaps is above a threshold value, the 'tug of war' between the forces of adhesion of graphene-graphene and graphene-substrate, flexural strain of folded region and carbon-carbon (C-C) covalent bonds favor the self-tearing and self-peeling off process. As the detailed information regarding the atomic scale mechanism involved in the process remains not fully understood, we carried out atomistic reactive molecular dynamics simulations to address some features of the process. We show that large thermal fluctuations can prevent the process by increasing the probability of chemical reactions between carbon dangling bonds of adjacent graphene layers. The effects of the strength of attraction between graphene and the substrate on the ribbon growth velocities at the early stages of the phenomenon were also investigated. Structures with initial armchair crack-edges were observed to form more uniform cuts than those having initial zigzag ones. Our results are of importance to help set up new experiments on this phenomenon, especially with samples with nanoscale sized cuts.},
note = {preprint arXiv:1808.08872},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
2019
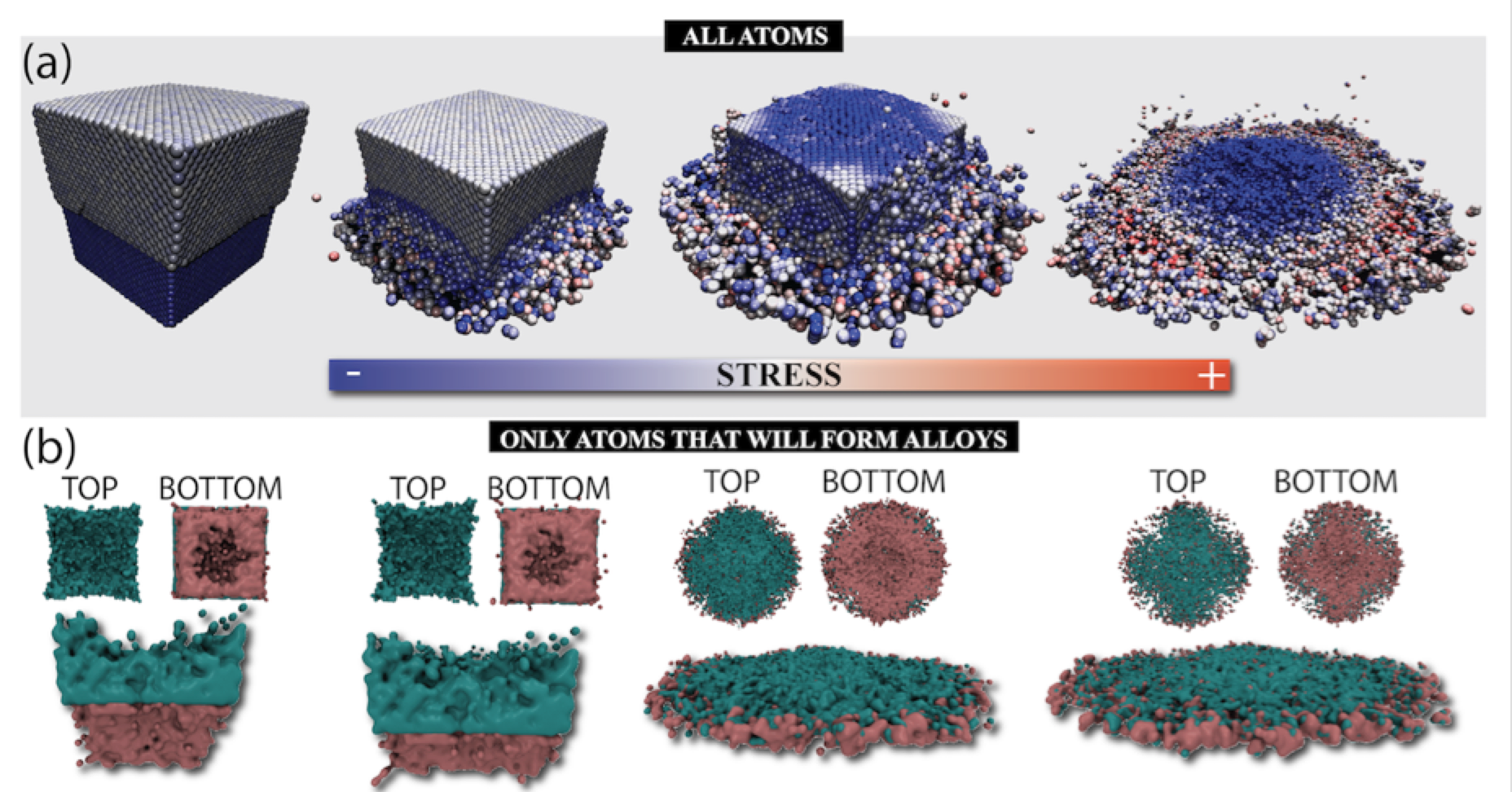
Malviya, Kirtman D; Oliveira, Eliezer F; Autreto, Pedro A S; Ajayan, Pulickel M; Galvao, D S; Tiwary, Candra S; Chattopadhyay, Kumanio
Mixing the immiscible through high-velocity mechanical impacts: an experimental and theoretical study Journal Article
In: Journal of Physics D: Applied Physics, vol. 52, no. 44, pp. 445304, 2019.
Abstract | Links | BibTeX | Tags: Mechanical Properties, Metal, Molecular Dynamics
@article{Malviya2019,
title = {Mixing the immiscible through high-velocity mechanical impacts: an experimental and theoretical study},
author = {Malviya, Kirtman D and Oliveira, Eliezer F and Autreto, Pedro A S and Ajayan, Pulickel M and Galvao, D S and Tiwary, Candra S and Chattopadhyay, Kumanio},
url = {https://iopscience.iop.org/article/10.1088/1361-6463/ab36d1/meta},
doi = {10.1088/1361-6463/ab36d1},
year = {2019},
date = {2019-08-20},
journal = {Journal of Physics D: Applied Physics},
volume = {52},
number = {44},
pages = {445304},
abstract = {In two-component metallic systems, thermodynamic immiscibility leads to phase separation
such as in two-phase eutectic compositional alloys. The limit of the immiscibility of
component elements under non-equilibrium conditions have been explored, but achieving
complete miscibility and formation of single phase microstructures in eutectic alloys would
be unprecedented. Here we report that during low-temperature ball milling that provides high
energy impact, complete mixing of phases can occur in immiscible Ag-Cu eutectic alloys.
From combined theoretical and experimental studies, we show that impact can produce solid
solutions of Ag-Cu nanoparticles of eutectic composition. Our results show that phase
diagrams of low dimensional materials under non-equilibrium conditions remain unexplored
and could lead to new alloy microstructures drastically different from their bulk counterparts.},
keywords = {Mechanical Properties, Metal, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
such as in two-phase eutectic compositional alloys. The limit of the immiscibility of
component elements under non-equilibrium conditions have been explored, but achieving
complete miscibility and formation of single phase microstructures in eutectic alloys would
be unprecedented. Here we report that during low-temperature ball milling that provides high
energy impact, complete mixing of phases can occur in immiscible Ag-Cu eutectic alloys.
From combined theoretical and experimental studies, we show that impact can produce solid
solutions of Ag-Cu nanoparticles of eutectic composition. Our results show that phase
diagrams of low dimensional materials under non-equilibrium conditions remain unexplored
and could lead to new alloy microstructures drastically different from their bulk counterparts.
de Sousa, Jose Moreira; Autreto, Pedro da Silva; Galvao, Douglas Soares
Hydrogenation Dynamics Process of Single-wall Carbon Nanotube Twisted (under review) Journal Article
In: 2019.
BibTeX | Tags: Carbon Nanotubes, Hydrogenation, Mechanical Properties, Molecular Dynamics
@article{deSousa2019d,
title = {Hydrogenation Dynamics Process of Single-wall Carbon Nanotube Twisted (under review)},
author = {de Sousa, Jose Moreira and Autreto, Pedro da Silva and Galvao, Douglas Soares},
year = {2019},
date = {2019-07-15},
keywords = {Carbon Nanotubes, Hydrogenation, Mechanical Properties, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
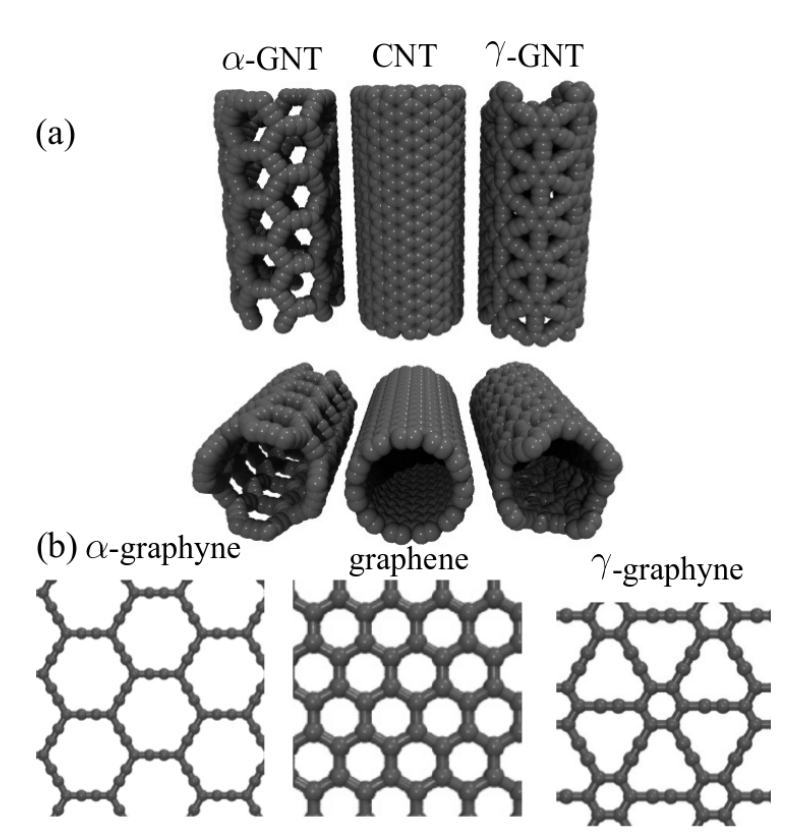
JM; Sousa, Bizao
Elastic Properties of Graphyne-based Nanotubes Online
2019, (ArXiv preprint.).
Abstract | Links | BibTeX | Tags: DFT, Graphynes, Molecular Dynamics, Nanotubes
@online{deSousa2019b,
title = {Elastic Properties of Graphyne-based Nanotubes},
author = {de Sousa, JM; , Bizao, RA; Sousa Filho, VP; Aguiar, AL; Coluci, VR; Pugno, NM; Girao, EC; Souza Filho, AG; Galvao, DS},
url = {https://arxiv.org/pdf/1905.02104.pdf},
year = {2019},
date = {2019-04-07},
abstract = {Graphyne nanotubes (GNTs) are nanostructures obtained from rolled up graphyne sheets,
in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes
are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to
conventional CNTs, GNTs can present different chiralities and electronic properties. Because
of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their
mechanical properties. In this work, we studied the mechanical response of GNTs under
tensile stress using fully atomistic molecular dynamics simulations and density functional
theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs
at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller
ultimate strength and Young’s modulus values. This is a consequence of the combined
effects of the existence of triple bonds and increased porosity/flexibility due to the presence
of acetylenic groups.},
note = {ArXiv preprint.},
keywords = {DFT, Graphynes, Molecular Dynamics, Nanotubes},
pubstate = {published},
tppubtype = {online}
}
in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes
are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to
conventional CNTs, GNTs can present different chiralities and electronic properties. Because
of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their
mechanical properties. In this work, we studied the mechanical response of GNTs under
tensile stress using fully atomistic molecular dynamics simulations and density functional
theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs
at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller
ultimate strength and Young’s modulus values. This is a consequence of the combined
effects of the existence of triple bonds and increased porosity/flexibility due to the presence
of acetylenic groups.
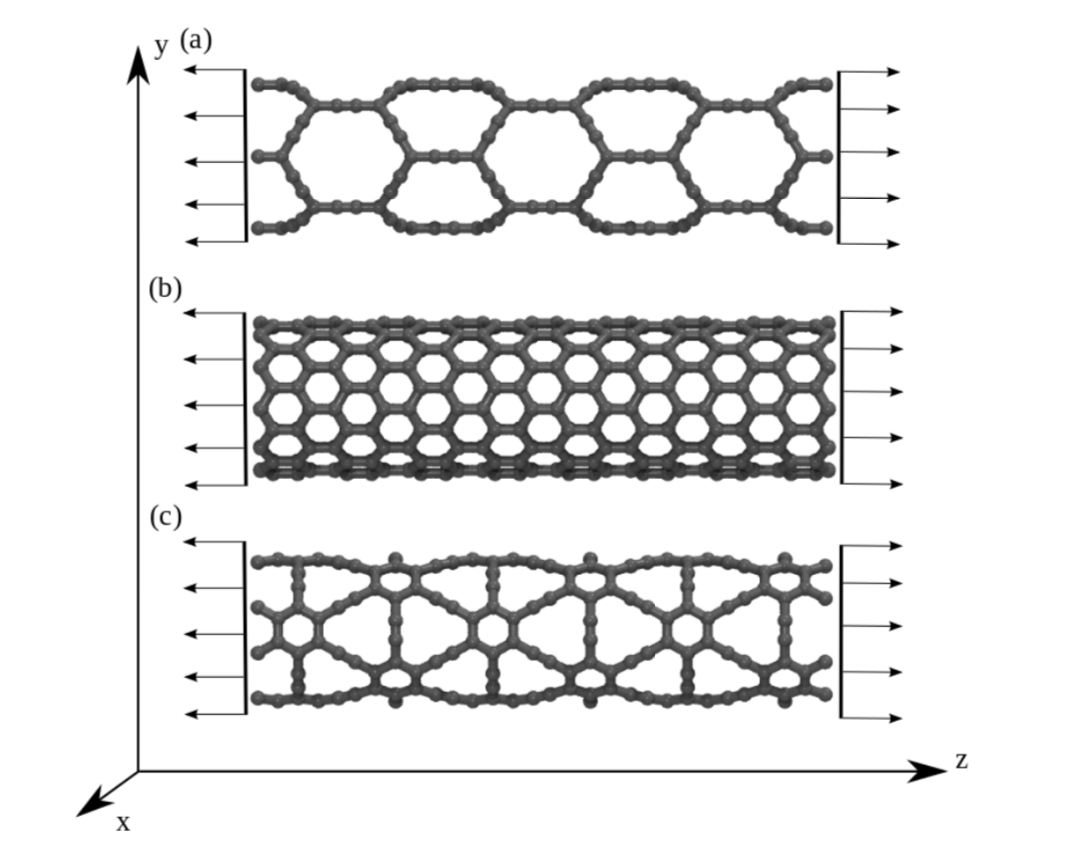
JM; Sousa, Bizao
Elastic Properties of Graphyne-Based Nanotubes Journal Article
In: Computational Materials Science, vol. 170, pp. 109153, 2019.
Abstract | Links | BibTeX | Tags: DFT, Graphynes, Molecular Dynamics, Nanotubes
@article{deSousa2019c,
title = {Elastic Properties of Graphyne-Based Nanotubes},
author = {de Sousa, JM; , Bizao, RA; Sousa Filho, VP; Aguiar, AL; Coluci, VR; Pugno, NM; Girao, EC; Souza Filho, AG; Galvao, DS},
url = {https://www.sciencedirect.com/science/article/pii/S0927025619304525?dgcid=coauthor#s0040},
doi = {10.1016/j.commatsci.2019.109153},
year = {2019},
date = {2019-04-03},
journal = {Computational Materials Science},
volume = {170},
pages = {109153},
abstract = {Graphyne nanotubes (GNTs) are nanostructures obtained from rolled up graphyne sheets, in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to conventional CNTs, GNTs can present different chiralities and electronic properties. Because of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their mechanical properties. In this work, we studied the mechanical response of GNTs under tensile stress using fully atomistic molecular dynamics simulations and density functional theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller ultimate strength and Young’s modulus values. This is a consequence of the combined effects of the existence of triple bonds and increased porosity/flexibility due to the presence of acetylenic groups.},
keywords = {DFT, Graphynes, Molecular Dynamics, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
Fonseca, AF; Dantas, SO; Galvao, DS; Zhang, D; Sinnott, SB
The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review) Journal Article
In: 2019.
BibTeX | Tags: C60, Graphene, Molecular Dynamics
@article{Fonseca2019d,
title = {The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review)},
author = {Fonseca, AF and Dantas, SO and Galvao, DS and Zhang, D and Sinnott, SB},
year = {2019},
date = {2019-04-03},
keywords = {C60, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Arpan; Gumaste Rout, Anurag; Pandey
Bio-inspired Aluminum Composite reinforced with Soft polymer with enhanced strength and plasticity (under review) Journal Article
In: 2019.
BibTeX | Tags: Metal, Molecular Dynamics, Polymers
@article{Rout2019,
title = {Bio-inspired Aluminum Composite reinforced with Soft polymer with enhanced strength and plasticity (under review)},
author = {Rout, Arpan; Gumaste, Anurag; Pandey, Praful; Oliveira, Eliezer; Demiss,
Solomon; P., Mahesh; Bhatt, Chintan; Raphael, Kiran; Ayyagari, Ravi; Autreto, Pedro;
Palit, Mithun; Femi, Olu Emmanuel; Galvao, Douglas; Arora, Amit; Tiwary, Chandra},
year = {2019},
date = {2019-03-30},
keywords = {Metal, Molecular Dynamics, Polymers},
pubstate = {published},
tppubtype = {article}
}
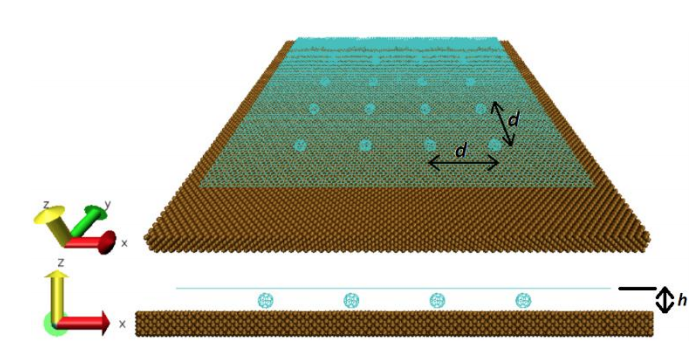
AF; Dantas Fonseca, SO; Galvao
The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study Online
2019, (ArXiv preprint).
Abstract | Links | BibTeX | Tags: C60, Graphene, Molecular Dynamics
@online{Fonseca2019b,
title = {The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study},
author = {Fonseca, AF; Dantas, SO; Galvao, DS; Zhang, D; Sinnott SB},
url = {https://arxiv.org/pdf/1904.09871.pdf},
year = {2019},
date = {2019-03-22},
abstract = {Two experimental studies reported the spontaneous formation of amorphous and crystalline
structures of C60 intercalated between graphene and a substrate. They observed interesting
phenomena ranging from reaction between C60 molecules under graphene to graphene
sagging between the molecules and control of strain in graphene. Motivated by these works,
we performed fully atomistic reactive molecular dynamics simulations to study the formation
and thermal stability of graphene wrinkles as well as graphene attachment to and detachment
from the substrate when graphene is laid over a previously distributed array of C60 molecules
on a copper substrate at different values of temperature. As graphene compresses the C60
molecules against the substrate, and graphene attachment to the substrate between C60s
(“C60S” stands for plural of C60) depends on the height of graphene wrinkles, configurations
with both frozen and non-frozen C60s structures were investigated in order to verify the
experimental result of stable sagged graphene when the distance between C60s is about 4 nm
and height of graphene wrinkles is about 0.8 nm. Below the distance of 4 nm between C60s,
graphene becomes locally suspended and less strained. We show that this happens when C60s
are allowed to deform under the compressive action of graphene. If we keep the C60s frozen,
spontaneous “blanketing” of graphene happens only when the distance between them are
equal or above 7 nm. Both above results for the existence of stable sagged graphene for C60
distances of 4 or 7 nm are shown to agree with a mechanical model relating the rigidity of
graphene to the energy of graphene-substrate adhesion. Although the studies of intercalation
of molecules on interfaces formed by graphene-substrate are motivated by finding out ways to
control wrinkling and strain in graphene, our work reveals the shape and structure of
intercalated molecules and the role of stability and wrinkling on final structure of graphene.
In particular, this study might help the development of 2D confined nanoreactors that are
considered in literature to be the next advanced step on chemical reactions.},
note = {ArXiv preprint},
keywords = {C60, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}
structures of C60 intercalated between graphene and a substrate. They observed interesting
phenomena ranging from reaction between C60 molecules under graphene to graphene
sagging between the molecules and control of strain in graphene. Motivated by these works,
we performed fully atomistic reactive molecular dynamics simulations to study the formation
and thermal stability of graphene wrinkles as well as graphene attachment to and detachment
from the substrate when graphene is laid over a previously distributed array of C60 molecules
on a copper substrate at different values of temperature. As graphene compresses the C60
molecules against the substrate, and graphene attachment to the substrate between C60s
(“C60S” stands for plural of C60) depends on the height of graphene wrinkles, configurations
with both frozen and non-frozen C60s structures were investigated in order to verify the
experimental result of stable sagged graphene when the distance between C60s is about 4 nm
and height of graphene wrinkles is about 0.8 nm. Below the distance of 4 nm between C60s,
graphene becomes locally suspended and less strained. We show that this happens when C60s
are allowed to deform under the compressive action of graphene. If we keep the C60s frozen,
spontaneous “blanketing” of graphene happens only when the distance between them are
equal or above 7 nm. Both above results for the existence of stable sagged graphene for C60
distances of 4 or 7 nm are shown to agree with a mechanical model relating the rigidity of
graphene to the energy of graphene-substrate adhesion. Although the studies of intercalation
of molecules on interfaces formed by graphene-substrate are motivated by finding out ways to
control wrinkling and strain in graphene, our work reveals the shape and structure of
intercalated molecules and the role of stability and wrinkling on final structure of graphene.
In particular, this study might help the development of 2D confined nanoreactors that are
considered in literature to be the next advanced step on chemical reactions.
AF; Dantas Fonseca, SO; Galvao
The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review) Journal Article
In: 2019.
BibTeX | Tags: C60, Graphene, Molecular Dynamics
@article{Fonseca2019c,
title = {The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review)},
author = {Fonseca, AF; Dantas, SO; Galvao, DS; Zhang, D; Sinnott SB},
year = {2019},
date = {2019-03-15},
keywords = {C60, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
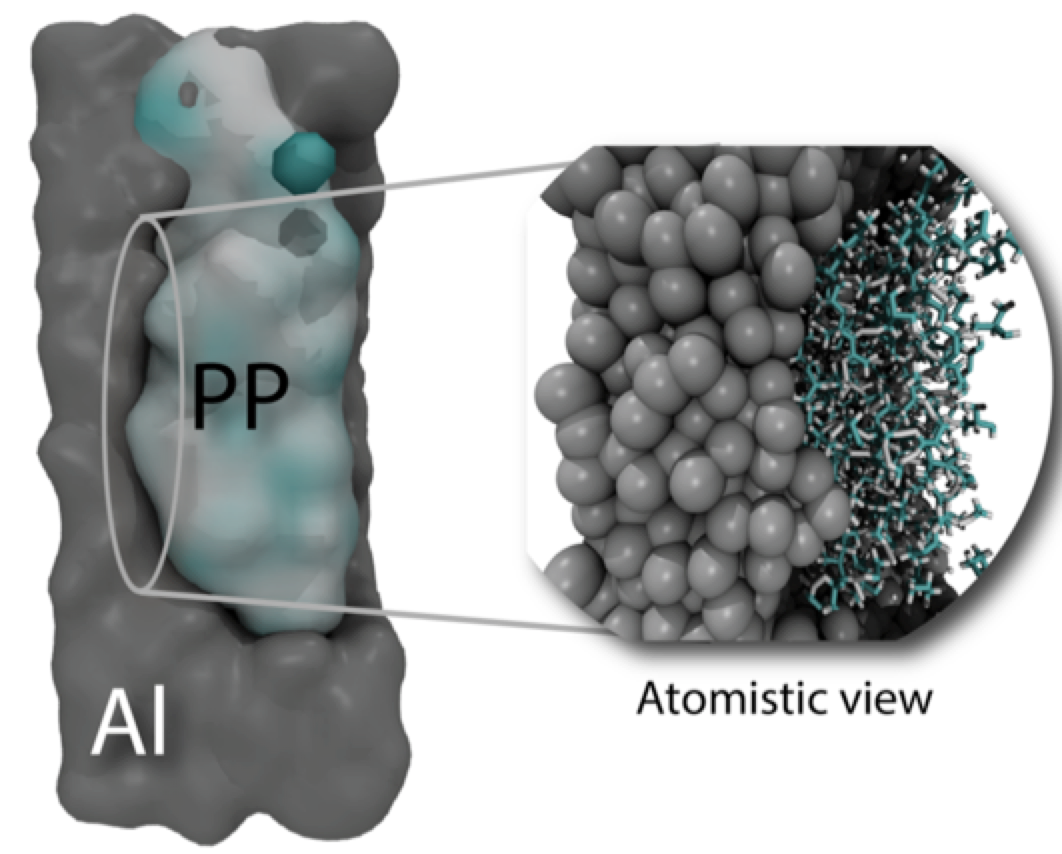
Routa, Arpan; Pandeyb, Praful; Oliveira, Eliezer Fernando; da Silva Autreto, Pedro Alves; Gumastea, Anurag; Singha, Amit; Galvao, Douglas Soares; Aroraa, Amit; Tiwary, Chandra Sekhar
Atomically locked interfaces of metal (Aluminum) and Polymer (Polypropylene) using mechanical friction Journal Article
In: Polymer, vol. 169, pp. 148-153, 2019.
Abstract | BibTeX | Tags: Composites, Metal, Molecular Dynamics, Polymers
@article{Routa2019,
title = {Atomically locked interfaces of metal (Aluminum) and Polymer (Polypropylene) using mechanical friction},
author = {Arpan Routa and Praful Pandeyb and Eliezer Fernando Oliveira and Pedro Alves da Silva Autreto and Anurag Gumastea and Amit Singha and Douglas Soares Galvao and Amit Aroraa and Chandra Sekhar Tiwary},
year = {2019},
date = {2019-02-23},
journal = {Polymer},
volume = {169},
pages = {148-153},
abstract = {Joining different parts is one of a crucial component of designing/engineering of materials. The current energy, low efficiency weight automotive and aerospace components commonly consist of different class of materials, such as metal, polymer, and ceramics, etc. Joining these components remains a challenge. Here, we demonstrate joining of metal (aluminum) and polymer (PP) using mechanical friction. The detailed characterisation demonstrates that atomically locked interfaces are formed in such joining without the presence of any chemical bond at the interfaces. The waterproof and strong interface is formed in such process. Fully atomistic molecular dynamics simulations were also carried out to provide further insights on these mechanisms.},
keywords = {Composites, Metal, Molecular Dynamics, Polymers},
pubstate = {published},
tppubtype = {article}
}
Eliezer F; Autreto Oliveira, Pedro AS; Woellner
On the mechanical properties of protomene: A theoretical investigation Journal Article
In: Computational Materials Science, vol. 161, pp. 190-198, 2019.
Abstract | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, protomene
@article{Oliveira2019c,
title = {On the mechanical properties of protomene: A theoretical investigation},
author = {Oliveira, Eliezer F; Autreto, Pedro AS; Woellner, Cristiano F; Galvao, Douglas S},
year = {2019},
date = {2019-02-07},
journal = {Computational Materials Science},
volume = {161},
pages = {190-198},
abstract = {We report a detailed study through fully atomistic molecular dynamics simulations and DFT calculations on the mechanical properties of protomene. Protomene is a new carbon allotrope composed of a mixture of sp2 and sp3 hybridized states. Our results indicate that protomene presents an anisotropic behavior about tensile deformations. At room temperature, protomene presents an ultimate strength of ~100 GPa and Young's modulus of ~600 GPa, lower than the same for other carbon allotropes. Despite that, protomente presents the highest ultimate strain along the z-direction (~ 24.7%). Our results also show that stretching the protomene along the z-direction or heating it can induce a semiconductor-metallic phase transition, due to a high amount of sp3 bonds that are converted to sp2 ones.},
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, protomene},
pubstate = {published},
tppubtype = {article}
}
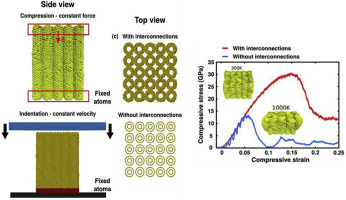
Sanjit; Ozden Bhowmick, Sehmus; Bizão
High temperature quasistatic and dynamic mechanical behavior of interconnected 3D carbon nanotube structures Journal Article
In: Carbon, vol. 142, pp. 291-299, 2019.
Abstract | Links | BibTeX | Tags: CNT, Fracture, Mechanical Properties, Molecular Dynamics
@article{Bhowmick2019,
title = {High temperature quasistatic and dynamic mechanical behavior of interconnected 3D carbon nanotube structures},
author = {Bhowmick, Sanjit; Ozden, Sehmus; Bizão, Rafael A; Machado, Leonardo Dantas; Asif, SA Syed; Pugno, Nicola M; Galvao, Douglas S; Tiwary, Chandra Sekhar; Ajayan, PM},
url = {https://www.sciencedirect.com/science/article/pii/S0008622318308911},
doi = {10.1016/j.carbon.2018.09.075},
year = {2019},
date = {2019-02-01},
journal = {Carbon},
volume = {142},
pages = {291-299},
abstract = {Carbon nanotubes (CNTs) are one of the most appealing materials in recent history for both research and commercial interest because of their outstanding physical, chemical, and electrical properties. This is particularly true for 3D arrangements of CNTs which enable their use in larger scale devices and structures. In this paper, the effect of temperature on the quasistatic and dynamic deformation behavior of 3D CNT structures is presented for the first time. An in situ high-temperature nanomechanical instrument was used inside an SEM at high vacuum to investigate mechanical properties of covalently interconnected CNT porous structures in a wide range of temperature. An irreversible bucking at the base of pillar samples was found as a major mode of deformation at room and elevated temperatures. It has been observed that elastic modulus and critical load to first buckle formation decrease progressively with increasing temperature from 25 °C to 750 °C. To understand fatigue resistance, pillars made from this unique structure were compressed to 100 cycles at room temperature and 750 °C. While the structure showed remarkable resistance to fatigue at room temperature, high temperature significantly lowers fatigue resistance. Molecular dynamics (MD) simulation of compression highlights the critical role played by covalent interconnections which prevent localized bending and improve mechanical properties.},
keywords = {CNT, Fracture, Mechanical Properties, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
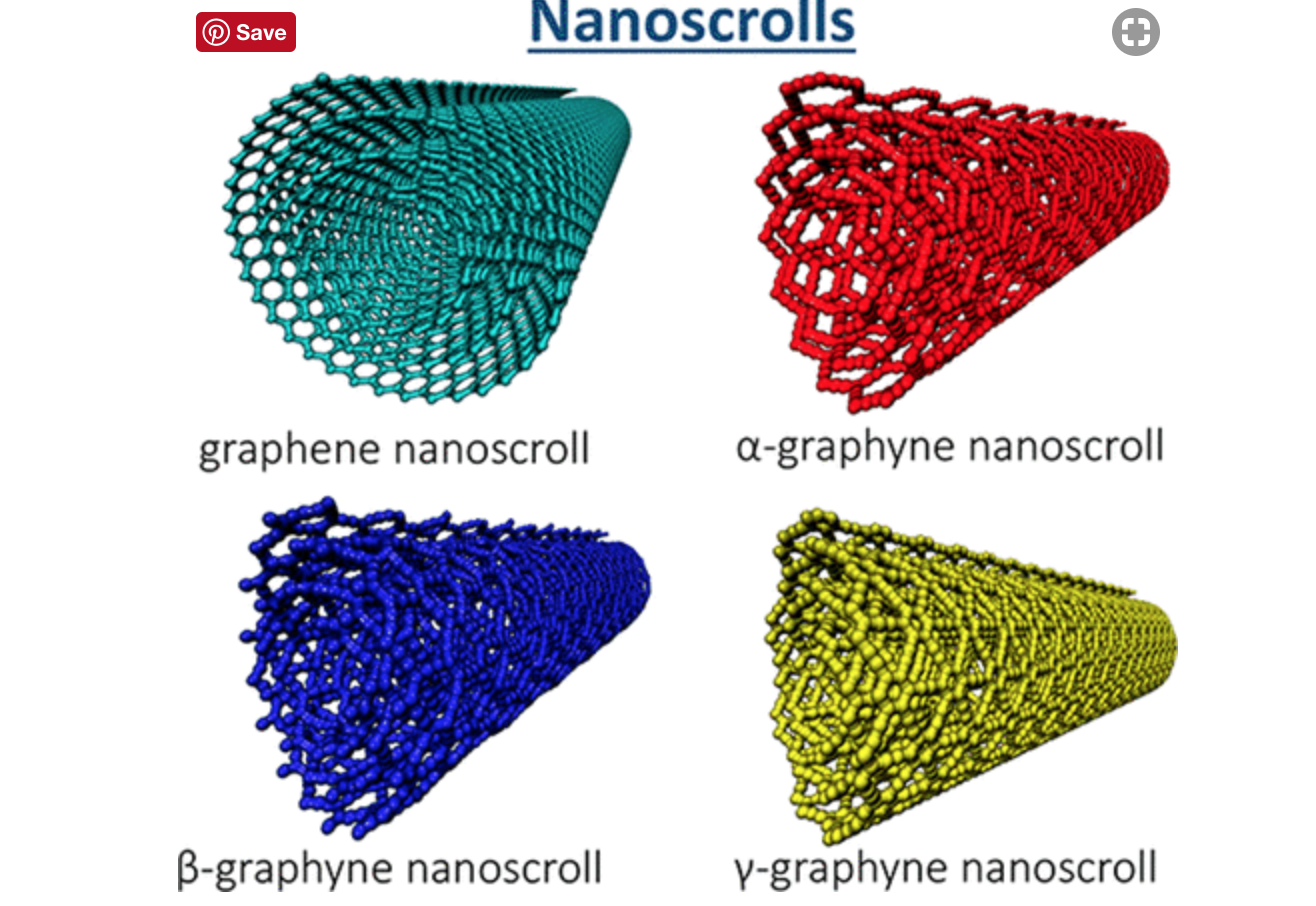
Solis, Daniel; Damasceno Borges, Daiane; Woellner, Cristiano; Galvao, Douglas
Structural and Thermal Stability of Graphyne and Graphdiyne Nanoscroll Structures (invited paper) Journal Article
In: ACS Applied Materials and Interfaces, vol. 11, pp. 2670−2676, 2019.
Abstract | Links | BibTeX | Tags: graphdiynes, Graphynes, Molecular Dynamics, Scrolls
@article{Solis2019,
title = {Structural and Thermal Stability of Graphyne and Graphdiyne Nanoscroll Structures (invited paper)},
author = {Solis, Daniel and Damasceno Borges, Daiane and Woellner, Cristiano and Galvao,
Douglas},
url = {https://pubs.acs.org/doi/10.1021/acsami.8b03481},
doi = {10.1021/acsami.8b03481},
year = {2019},
date = {2019-01-23},
journal = {ACS Applied Materials and Interfaces},
volume = {11},
pages = {2670−2676},
abstract = {Graphynes and graphdiynes are generic names for families of two-dimensional carbon allotropes, where acetylenic groups connect benzenoid-like hexagonal rings, with the coexistence of sp and sp2 hybridized carbon atoms. The main differences between graphynes and graphdiynes are the number of acetylenic groups (one and two for graphynes and graphdiynes, respectively). Similarly to graphene nanoscrolls, graphyne and graphdiynes nanoscrolls are nanosized membranes rolled into papyrus-like structures. In this work we studied through molecular dynamics simulations, using reactive potentials, the structural and thermal (up to 1000 K) stability of α,β,γ-graphyne and α,β,γ-graphdiyne scrolls. Our results demonstrate that stable nanoscrolls can be created for all the structures studied here, although they are less stable than corresponding graphene scrolls. This can be elucidated as a result of the higher graphyne/graphdiyne structural porosity in relation to graphene, and as a consequence, the π–π stacking interactions decrease.},
keywords = {graphdiynes, Graphynes, Molecular Dynamics, Scrolls},
pubstate = {published},
tppubtype = {article}
}
de Sousa, JM; Aguiar, AL; Girao, EC; Fonseca, Alexandre F; AG Filho, Souza; Galvao, Douglas S
Mechanical Properties and Fracture Patterns of Pentagraphene Membranes (under review) Journal Article
In: 2019.
BibTeX | Tags: Fracture, Molecular Dynamics, pentagraphene
@article{deSousa2019,
title = {Mechanical Properties and Fracture Patterns of Pentagraphene Membranes (under review)},
author = {de Sousa, JM and Aguiar, AL and Girao, EC and Fonseca, Alexandre F and AG Filho, Souza and Galvao, Douglas S},
year = {2019},
date = {2019-01-05},
keywords = {Fracture, Molecular Dynamics, pentagraphene},
pubstate = {published},
tppubtype = {article}
}

Fonseca, Alexandre F.; Galvao, Douglas S.
Self-tearing and self-peeling of folded graphene nanoribbons Journal Article
In: Carbon, vol. 143, pp. 230-239, 2019.
Abstract | Links | BibTeX | Tags: Fracture, Graphene, Mechanical Properties, Molecular Dynamics
@article{Fonseca2019,
title = {Self-tearing and self-peeling of folded graphene nanoribbons},
author = {Alexandre F. Fonseca and Douglas S. Galvao},
url = {https://www.sciencedirect.com/science/article/pii/S0008622318310431},
doi = {10.1016/j.carbon.2018.11.020},
year = {2019},
date = {2019-01-05},
journal = {Carbon},
volume = {143},
pages = {230-239},
abstract = {A recent experimental study showed that an induced folded flap of graphene can spontaneously drive itself its tearing and peeling off a substrate, thus producing long, micrometer sized, regular trapezoidal-shaped folded graphene nanoribbons. As long as the size of the graphene flaps is above a threshold value, the “tug of war” between the forces of adhesion of graphene-graphene and graphene-substrate, flexural strain of folded region and carbon-carbon (C-C) covalent bonds favor the self-tearing and self-peeling off process. As the detailed information regarding the atomic scale mechanism involved in the process remains not fully understood, we carried out atomistic reactive molecular dynamics simulations to address some features of the process. We show that large thermal fluctuations can prevent the process by increasing the probability of chemical reactions between carbon dangling bonds of adjacent graphene layers. The effects of the strength of attraction between graphene and the substrate on the ribbon growth velocities at the early stages of the phenomenon were also investigated. Structures with initial armchair crack-edges were observed to form more uniform cuts than those having initial zigzag ones. Our results are of importance to help set up new experiments on this phenomenon, especially with samples with nanoscale sized cuts. },
keywords = {Fracture, Graphene, Mechanical Properties, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Sean P; Perim Collins, Eric; Daff
Idealized Carbon-Based Materials Exhibiting Record Deliverable Capacities for Vehicular Methane Storage Journal Article
In: The Journal of Physical Chemistry C, vol. 123, pp. 1050-1058, 2019.
Abstract | Links | BibTeX | Tags: Gas Storage, Molecular Dynamics, Monte Carlo, Schwarzites, Scrolls
@article{Collins2019,
title = {Idealized Carbon-Based Materials Exhibiting Record Deliverable Capacities for Vehicular Methane Storage},
author = {Collins, Sean P; Perim, Eric; Daff, Thomas D; Skaf, Munir S; Galvao, Douglas Soares; Woo, Tom K},
url = {https://pubs.acs.org/doi/abs/10.1021/acs.jpcc.8b09447},
doi = {10.1021/acs.jpcc.8b09447},
year = {2019},
date = {2019-01-05},
journal = {The Journal of Physical Chemistry C},
volume = {123},
pages = {1050-1058},
abstract = {Materials for vehicular methane storage have been extensively studied, although no suitable material has been found. In this work, we use molecular simulation to investigate three types of carbon-based materials, Schwarzites, layered graphenes, and carbon nanoscrolls, for use in vehicular methane storage under adsorption conditions of 65 bar and 298 K and desorption conditions of 5.8 bar and 358 K. Ten different Schwarzites were tested and found to have high adsorption with maximums at 273 VSTP/V, but middling deliverable capacities of no more than 131 VSTP/V. Layered graphene and graphene nanoscrolls were found to have extremely high CH4 adsorption capacities of 355 and 339 VSTP/V, respectively, when the interlayer distance was optimized to 11 Å. The deliverable capacities of perfectly layered graphene and graphene nanoscrolls were also found to be exceptional with values of 266 and 252 VSTP/V, respectively, with optimized interlayer distances. These values make idealized graphene and nanoscrolls the record holders for adsorption and deliverable capacities under vehicular methane storage conditions.},
keywords = {Gas Storage, Molecular Dynamics, Monte Carlo, Schwarzites, Scrolls},
pubstate = {published},
tppubtype = {article}
}
Owuor, Peter Samora; Inthong, Suchittra; Sajadi, Seyed Mohammad; Intawin, Pratthana; Chipara, Alin C.; Woellner, Cristiano F.; Sayed, Farheen N.; Tsang, Harvey H.; Stender, Anthony; Vajtai, Robert; Pengpat, Kamonpan; Eitssayeam, Sukum; Galvao, Douglas S.; Lou, Jun; Tiwary, Chandra Sekhar; Ajayan, Pulickel M.
Elastic and ‘transparent bone’ as an electrochemical separator Journal Article
In: Materials Chemistry Today, vol. 12, pp. 132-138, 2019.
Abstract | Links | BibTeX | Tags: biomaterials, Bone, Characterization, electrodes, Modeling, Molecular Dynamics
@article{Owuor2019,
title = {Elastic and ‘transparent bone’ as an electrochemical separator},
author = {Peter Samora Owuor and Suchittra Inthong and Seyed Mohammad Sajadi and Pratthana Intawin and Alin C. Chipara and Cristiano F. Woellner and Farheen N. Sayed and Harvey H. Tsang and Anthony Stender and Robert Vajtai and Kamonpan Pengpat and Sukum Eitssayeam and Douglas S. Galvao and Jun Lou and Chandra Sekhar Tiwary and Pulickel M. Ajayan},
url = {https://reader.elsevier.com/reader/sd/pii/S246851941830291X?token=B3C1F35B7DCEA8636EFB32B8D1D71EEC9852E58D0729A622DAFDF86C3EE65DF2A33E77CE7534A5D66D3854C396F69D1A},
doi = {10.1016/j.mtchem.2018.12.009},
year = {2019},
date = {2019-01-05},
journal = {Materials Chemistry Today},
volume = {12},
pages = {132-138},
abstract = {Organic matrix of bone mainly composed of collagen matrix serve as a crucial component for remarkable toughness and strength in bones. The porous collagen matrix can also serve as efficient template for various applications such as nanoparticles synthetic, catalysis or catalysis supports, electrochemical separator, filtration membrane and tissue engineering. However, fabricating collagen matrix from bones without degrading its morphological structure still remain a challenge. Here we present evidence of how ceramic crystals from a bone can be removed to fabricate a complete ‘transparent bone’ structure with improved porous and elasticity. We show that demineralization or selective etching using dilute acid (citric) can remove ceramics mineral nanoparticles without degrading the collagen matrix. The transparent bone collagen matrix is investigated as the separator in electrochemical supercapacitor with aqueous electrolyte where it shows better performance compared to conventional separators.},
keywords = {biomaterials, Bone, Characterization, electrodes, Modeling, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Eliezer F; Autreto Oliveira, Pedro AS; Woellner
Mechanical Properties of Protomene: A Molecular Dynamics Investigation Journal Article
In: MRS Advances, 2019.
Abstract | Links | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, protomene
@article{Oliveira2019,
title = {Mechanical Properties of Protomene: A Molecular Dynamics Investigation},
author = {Oliveira, Eliezer F; Autreto, Pedro AS; Woellner, Cristiano F; Galvao, Douglas S},
url = {www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-protomene-a-molecular-dynamics-investigation/CBAC89BDB5942E3353A5C00BD5D0D9CA},
doi = {10.1557/adv.2018.670},
year = {2019},
date = {2019-01-05},
journal = {MRS Advances},
abstract = {Recently, a new class of carbon allotrope called protomene was proposed. This new structure is composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3 carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now, its mechanical properties have not been investigated. In this work, we have investigated protomene mechanical behavior under tensile strain through fully atomistic reactive molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS code. At room temperature, our results show that the protomene is very stable and the obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical fracture.
},
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, protomene},
pubstate = {published},
tppubtype = {article}
}
Nakar, Dekel; Gordeev, Georgy; Machado, Leonardo D.; Popovitz-Biro, Ronit; Rechav, Katya; Oliveira, Eliezer F.; Kusch, Patryk; Jorio, Ado; Galvao, Douglas S.; Reich, Stephanie; Joselevich, Ernesto
Few-Wall Carbon Nanotube Coils (under review) Journal Article
In: 2019.
BibTeX | Tags: Carbon Nanotubes, Molecular Dynamics, Nanocoils, Raman
@article{Nakar2019,
title = {Few-Wall Carbon Nanotube Coils (under review)},
author = {Dekel Nakar and Georgy Gordeev and Leonardo D. Machado and Ronit Popovitz-Biro and Katya Rechav and Eliezer F. Oliveira and Patryk Kusch and Ado Jorio and Douglas S. Galvao and Stephanie Reich and Ernesto Joselevich},
year = {2019},
date = {2019-01-01},
keywords = {Carbon Nanotubes, Molecular Dynamics, Nanocoils, Raman},
pubstate = {published},
tppubtype = {article}
}
2018
Pedro AS Autreto Eliezer F Oliveira, Cristiano F Woellner
Mechanical Properties of Protomene: A Molecular Dynamics Investigation Online
2018, (preprint arXiv:1810.09924v1 ).
Abstract | Links | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, protomene
@online{Oliveira2018g,
title = {Mechanical Properties of Protomene: A Molecular Dynamics Investigation},
author = {Eliezer F Oliveira, Pedro AS Autreto, Cristiano F Woellner, Douglas S Galvao},
url = {https://arxiv.org/abs/1810.09924},
year = {2018},
date = {2018-10-23},
abstract = {Recently, a new class of carbon allotrope called protomene was proposed. This new structure is
composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3
carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations
have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now,
its mechanical properties have not been investigated. In this work, we have investigated
protomene mechanical behavior under tensile strain through fully atomistic reactive
molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS
code. At room temperature, our results show that the protomene is very stable and the
obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest
ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate
strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical
fracture.},
note = {preprint arXiv:1810.09924v1 },
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, protomene},
pubstate = {published},
tppubtype = {online}
}
composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3
carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations
have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now,
its mechanical properties have not been investigated. In this work, we have investigated
protomene mechanical behavior under tensile strain through fully atomistic reactive
molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS
code. At room temperature, our results show that the protomene is very stable and the
obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest
ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate
strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical
fracture.
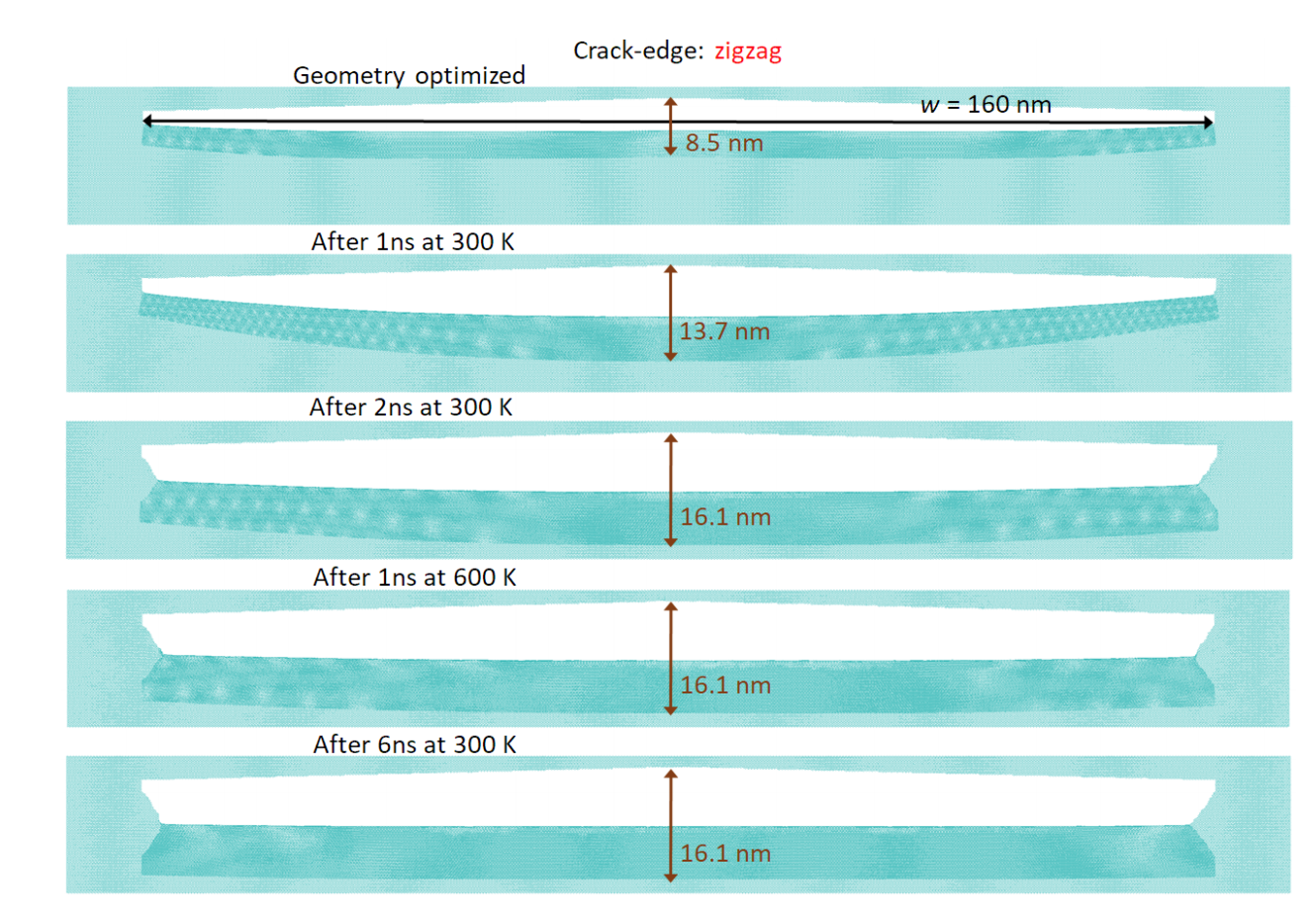
Alexandre F. Fonseca, Douglas S. Galvao
Self-tearing and self-peeling of folded graphene nanoribbons Online
2018, (preprint arXiv:1808.08872).
Abstract | Links | BibTeX | Tags: Fracture, graphene nanoribbons, Mechanical Properties, Molecular Dynamics
@online{Fonseca2018d,
title = { Self-tearing and self-peeling of folded graphene nanoribbons},
author = {Alexandre F. Fonseca, Douglas S. Galvao
},
url = {https://arxiv.org/abs/1808.08872},
year = {2018},
date = {2018-08-27},
abstract = {A recent experimental study showed that an induced folded flap of graphene can spontaneously drive itself its tearing and peeling off a substrate, thus producing long, micrometer sized, regular trapezoidal-shaped folded graphene nanoribbons. As long as the size of the graphene flaps is above a threshold value, the 'tug of war' between the forces of adhesion of graphene-graphene and graphene-substrate, flexural strain of folded region and carbon-carbon (C-C) covalent bonds favor the self-tearing and self-peeling off process. As the detailed information regarding the atomic scale mechanism involved in the process remains not fully understood, we carried out atomistic reactive molecular dynamics simulations to address some features of the process. We show that large thermal fluctuations can prevent the process by increasing the probability of chemical reactions between carbon dangling bonds of adjacent graphene layers. The effects of the strength of attraction between graphene and the substrate on the ribbon growth velocities at the early stages of the phenomenon were also investigated. Structures with initial armchair crack-edges were observed to form more uniform cuts than those having initial zigzag ones. Our results are of importance to help set up new experiments on this phenomenon, especially with samples with nanoscale sized cuts.},
note = {preprint arXiv:1808.08872},
keywords = {Fracture, graphene nanoribbons, Mechanical Properties, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}
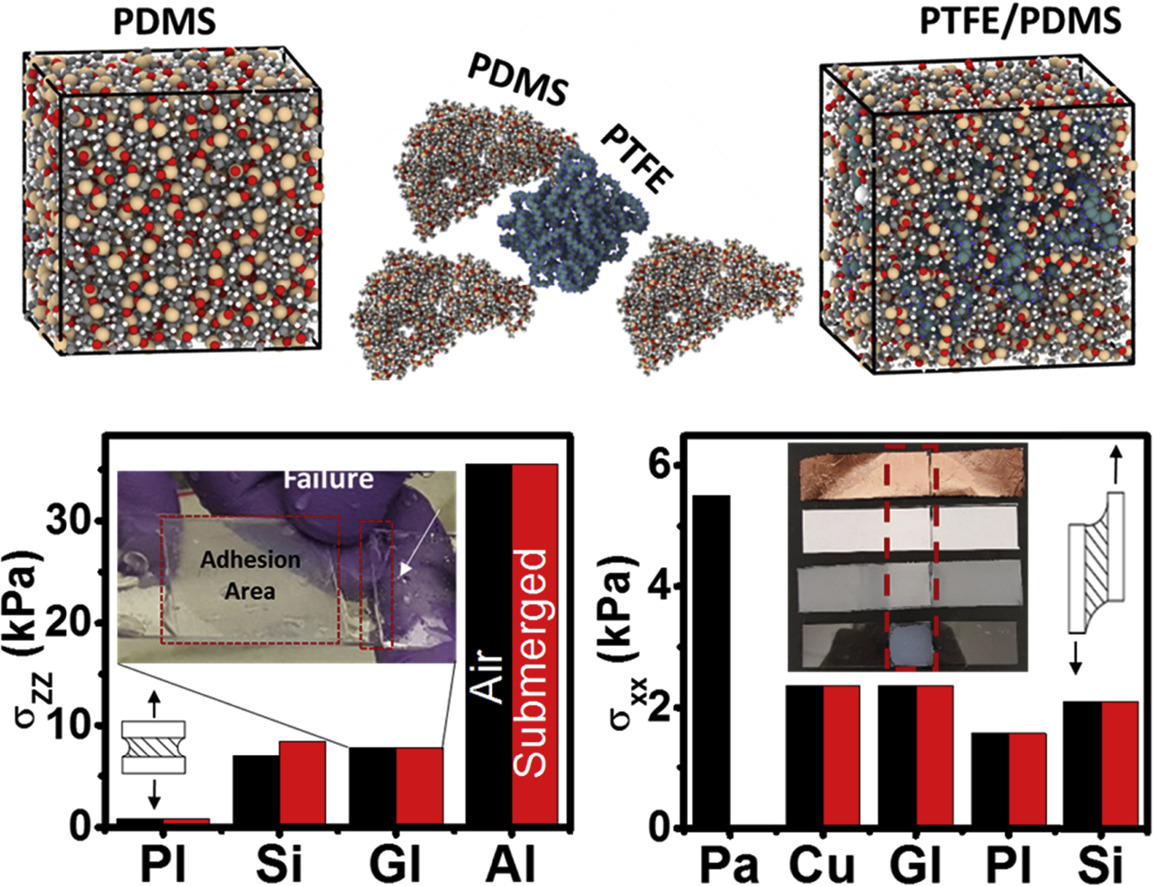
Chipara, A. C.; Tsafack, T.; Owuor, P. S.; Yeon, J.; Junkermeier, C. E.; van Duin, A. C. T.; Bhowmick, S.; Asif, S. A. S.; Radhakrishnan, S.; Park, J. H.; Brunetto, G.; Kaipparettu, B. A.; Galvão, D. S.; Chipara, M.; Lou, J.; Tsang, H. H.; Dubey, M.; Vajtai, R.; Tiwary, C. S.; Ajayan, P. M.
Underwater Adhesive using Solid–liquid Polymer Mixes Journal Article
In: Materials Today Chemistry, vol. 9, pp. 149-157, 2018.
Abstract | Links | BibTeX | Tags: Adhesives, DFT, Molecular Dynamics, Polymer
@article{Chipara2018,
title = {Underwater Adhesive using Solid–liquid Polymer Mixes},
author = {A.C. Chipara and T. Tsafack and P.S. Owuor and J. Yeon and C.E. Junkermeier and A.C.T. van Duin and S. Bhowmick and S.A.S. Asif and S. Radhakrishnan and J.H. Park and G. Brunetto and B.A. Kaipparettu and D.S. Galvão and M. Chipara and J. Lou and H.H. Tsang and M. Dubey and R. Vajtai and C.S. Tiwary and P.M. Ajayan},
url = {https://www.sciencedirect.com/science/article/pii/S2468519418301423#appsec1},
doi = {10.1016/j.mtchem.2018.07.002},
year = {2018},
date = {2018-08-08},
journal = {Materials Today Chemistry},
volume = {9},
pages = {149-157},
abstract = {Instantaneous adhesion between different materials is a requirement for several applications ranging from electronics to biomedicine. Approaches such as surface patterning, chemical cross-linking, surface modification, and chemical synthesis have been adopted to generate temporary adhesion between various materials and surfaces. Because of the lack of curing times, temporary adhesives are instantaneous, a useful property for specific applications that need quick bonding. However, to this day, temporary adhesives have been mainly demonstrated under dry conditions and do not work well in submerged or humid environments. Furthermore, most rely on chemical bonds resulting from strong interactions with the substrate such as acrylate based. This work demonstrates the synthesis of a universal amphibious adhesive solely by combining solid polytetrafluoroethylene (PTFE) and liquid polydimethylsiloxane (PDMS) polymers. While the dipole-dipole interactions are induced by a large electronegativity difference between fluorine atoms in PTFE and hydrogen atoms in PDMS, strong surface wetting allows the proposed adhesive to fully coat both substrates and PTFE particles, thereby maximizing the interfacial chemistry. The two-phase solid–liquid polymer system displays adhesive characteristics applicable both in air and water, and enables joining of a wide range of similar and dissimilar materials (glasses, metals, ceramics, papers, and biomaterials). The adhesive exhibits excellent mechanical properties for the joints between various surfaces as observed in lap shear testing, T-peel testing, and tensile testing. The proposed biocompatible adhesive can also be reused multiple times in different dry and wet environments. Additionally, we have developed a new reactive force field parameterization and used it in our molecular dynamics simulations to validate the adhesive nature of the mixed polymer system with different surfaces. This simple amphibious adhesive could meet the need for a universal glue that performs well with a number of materials for a wide range of conditions.},
keywords = {Adhesives, DFT, Molecular Dynamics, Polymer},
pubstate = {published},
tppubtype = {article}
}
Oliveira, Eliezer F.; Autreto, Pedro A. S.; Woellner, Cristiano F.; Galvao, Douglas S.
On the mechanical properties of novamene: a fully atomistic molecular dynamics and DFT investigation Journal Article
In: Carbon, vol. 139, pp. 782-788, 2018.
Abstract | Links | BibTeX | Tags: carbon allotropes, DFT, Molecular Dynamics, novamenes
@article{Oliveira2018e,
title = {On the mechanical properties of novamene: a fully atomistic molecular dynamics and DFT investigation},
author = {Eliezer F. Oliveira and Pedro A. S. Autreto and Cristiano F. Woellner and Douglas S. Galvao},
url = {https://www.sciencedirect.com/science/article/pii/S0008622318306882?via%3Dihub#appsec1},
doi = {10.1016/j.carbon.2018.07.038},
year = {2018},
date = {2018-07-19},
journal = {Carbon},
volume = {139},
pages = {782-788},
abstract = {We have investigated through fully atomistic reactive molecular dynamics and density functional theory simulations, the mechanical properties and fracture dynamics of single-ringed novamene (1R-novamene), a new 3D carbon allotrope structure recently proposed. Our results showed that 1R-novamene is an anisotropic structure with relation to tensile deformation. Although 1R-novamente shares some mechanical features with other carbon allotropes, it also exhibits distinct ones, such as, extensive structural reconstructions. 1R-novamene presents ultimate strength (∼100 GPa) values lower than other carbon allotropes, but it has the highest ultimate strain along the z-direction (∼22.5%). Although the Young's modulus (∼600 GPa) and ultimate strength values are smaller than for other carbon allotropes, they still outperform other materials, such as for example silicon, steel or titanium alloys. With relation to the fracture dynamics, 1R-novamene is again anisotropic with the fracture/crack propagation originating from deformed heptagons and pentagons for x and y directions and broken sp3 bonds connecting structural planes. Another interesting feature is the formation of multiple and long carbon linear chains in the final fracture stages.},
keywords = {carbon allotropes, DFT, Molecular Dynamics, novamenes},
pubstate = {published},
tppubtype = {article}
}
Gautam, Chandkiram; Chakravarty, Dibyendu; Woellner, Cristiano F.; Mishra, Vijay Kumar; Ahamad, Naseer; Gautam, Amarendra; Ozden, Sehmus; Jose, Sujin; Biradar, Santosh Kumar; Vajtai, Robert; Trivedi, Ritu; Tiwary, Chandra Sekhar; Galvao, Douglas S.; Ajayan, P. M.
Synthesis and 3D Interconnected Nanostructured h-BN-Based Biocomposites by Low-Temperature Plasma Sintering: Bone Regeneration Applications Journal Article
In: ACS Omega, vol. 3, no. 6, pp. 6013–6021, 2018.
Abstract | Links | BibTeX | Tags: BN, Composites, Molecular Dynamics, sintering
@article{Gautam2018,
title = {Synthesis and 3D Interconnected Nanostructured h-BN-Based Biocomposites by Low-Temperature Plasma Sintering: Bone Regeneration Applications},
author = {Chandkiram Gautam and Dibyendu Chakravarty and Cristiano F. Woellner and Vijay Kumar Mishra and Naseer Ahamad and Amarendra Gautam and Sehmus Ozden and Sujin Jose and Santosh Kumar Biradar and Robert Vajtai and Ritu Trivedi and Chandra Sekhar Tiwary and Douglas S. Galvao and P.M. Ajayan},
url = {https://pubs.acs.org/doi/abs/10.1021/acsomega.8b00707},
doi = {10.1021/acsomega.8b00707},
year = {2018},
date = {2018-06-05},
journal = {ACS Omega},
volume = {3},
number = {6},
pages = {6013–6021},
abstract = {Recent advances and demands in biomedical applications drive a large amount of research to synthesize easily scalable low-density, high-strength, and wear-resistant biomaterials. The chemical inertness with low density combined with high strength makes h-BN one of the promising materials for such application. In this work, three-dimensional hexagonal boron nitride (h-BN) interconnected with boron trioxide (B2O3) was prepared by easily scalable and energy efficient spark plasma sintering (SPS) process. The composite structure shows significant densification (1.6–1.9 g/cm3) and high surface area (0.97–14.5 m2/g) at an extremely low SPS temperature of 250 °C. A high compressive strength of 291 MPa with a reasonably good wear resistance was obtained for the composite structure. The formation of strong covalent bonds between h-BN and B2O3 was formulated and established by molecular dynamics simulation. The composite showed significant effect on cell viability/proliferation. It shows a high mineralized nodule formation over the control, which suggests its use as a possible osteogenic agent in bone formation.},
keywords = {BN, Composites, Molecular Dynamics, sintering},
pubstate = {published},
tppubtype = {article}
}
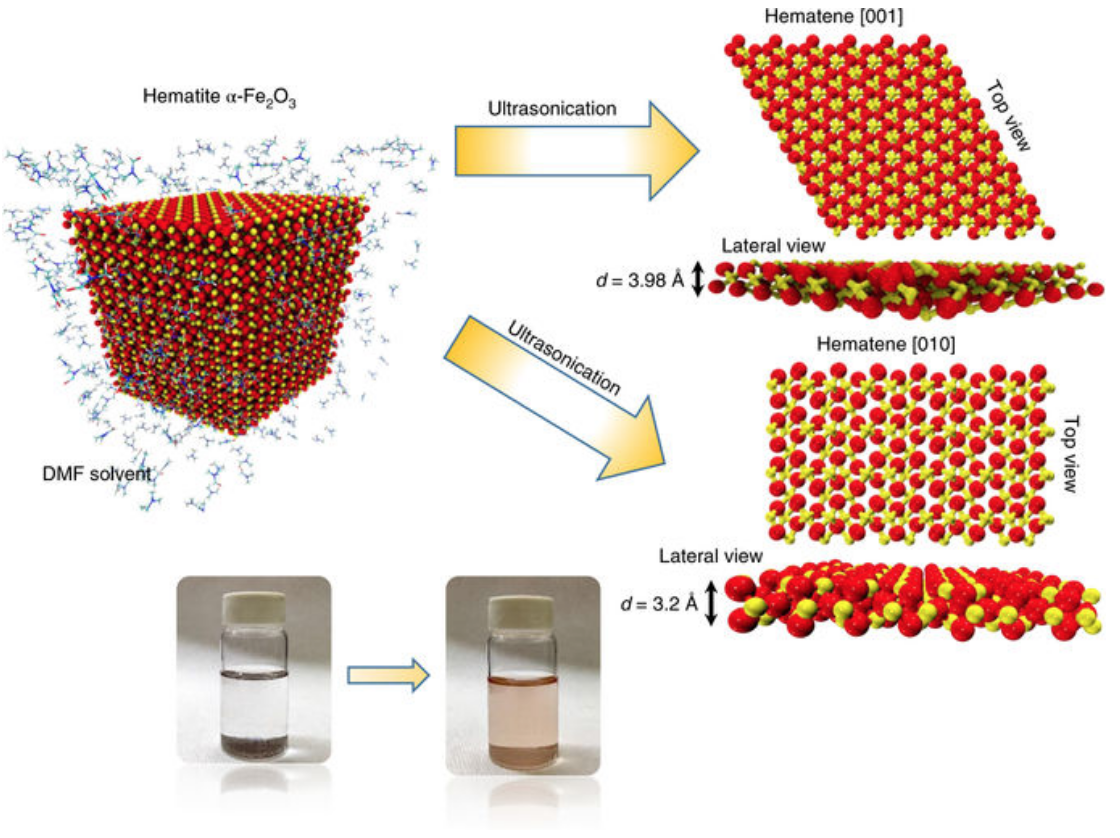
Balan, Aravind Puthirath; Radhakrishnan, Sruthi; Woellner, Cristiano F.; Sinha, Shyam K.; Deng, Liangzi; de los Reyes, Carlos; Rao, Manmadha; Paulose, Maggie; Neupane, Ram; Vajtai, Robert; Chu, Ching-Wu; Costin, Gelu; Galvao, Douglas S.; Marti, Angel A.; van Aken, Peter; Varghese, Oomman K; Tiwary, Chandra Sekhar; Anantharaman, M R; Ajayan, Pulickel M
Exfoliation of a non-van der Waals material from iron ore hematite Journal Article
In: Nature Nanotechnology, vol. 13, pp. 602–610, 2018.
Abstract | Links | BibTeX | Tags: DFT, Hematene, Molecular Dynamics, van der Waals solids
@article{Balan2018,
title = {Exfoliation of a non-van der Waals material from iron ore hematite},
author = {Aravind Puthirath Balan and Sruthi Radhakrishnan and Cristiano F. Woellner and Shyam K. Sinha and Liangzi Deng and Carlos de los Reyes and Manmadha Rao and Maggie Paulose and Ram Neupane and Robert Vajtai and Ching-Wu Chu and Gelu Costin and Douglas S. Galvao and Angel A. Marti and Peter van Aken and Oomman K Varghese and Chandra Sekhar Tiwary and M R Anantharaman and Pulickel M Ajayan
},
url = {https://www.nature.com/articles/s41565-018-0134-y},
year = {2018},
date = {2018-05-07},
journal = {Nature Nanotechnology},
volume = {13},
pages = {602--610},
abstract = {With the advent of graphene, the most studied of all two-dimensional materials, many inorganic analogues have been synthesized and are being exploited for novel applications. Several approaches have been used to obtain large-grain, high-quality materials. Naturally occurring ores, for example, are the best precursors for obtaining highly ordered and large-grain atomic layers by exfoliation. Here, we demonstrate a new two-dimensional material ‘hematene’ obtained from natural iron ore hematite (α-Fe2O3), which is isolated by means of liquid exfoliation. The two-dimensional morphology of hematene is confirmed by transmission electron microscopy. Magnetic measurements together with density functional theory calculations confirm the ferromagnetic order in hematene while its parent form exhibits antiferromagnetic order. When loaded on titania nanotube arrays, hematene exhibits enhanced visible light photocatalytic activity. Our study indicates that photogenerated electrons can be transferred from hematene to titania despite a band alignment unfavourable for charge transfer.},
keywords = {DFT, Hematene, Molecular Dynamics, van der Waals solids},
pubstate = {published},
tppubtype = {article}
}
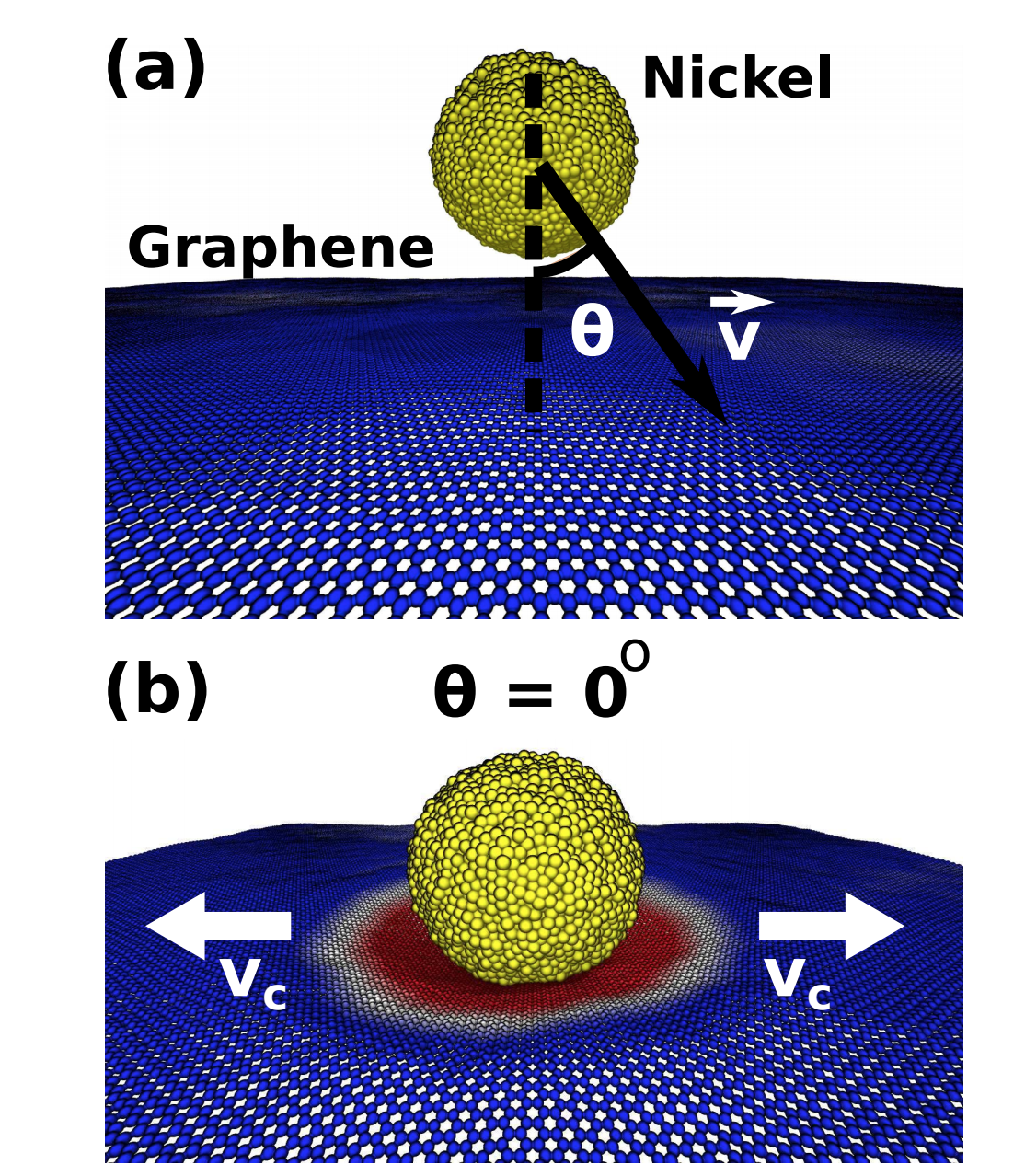
Bizao, Rafael A; Machado, Leonardo D; de Sousa, Jose M; Pugno, Nicola M; Galvao, Douglas S
Scale Effects on the Ballistic Penetration of Graphene Sheets Journal Article
In: Nature Scientific Reports, vol. 8, pp. 6750, 2018.
Abstract | Links | BibTeX | Tags: Fracture, Graphene, impact, Molecular Dynamics
@article{Bizao2018,
title = {Scale Effects on the Ballistic Penetration of Graphene Sheets},
author = {Bizao, Rafael A and Machado, Leonardo D and de Sousa, Jose M and Pugno, Nicola M and Galvao, Douglas S},
url = {https://www.nature.com/articles/s41598-018-25050-2},
doi = {doi:10.1038/s41598-018-25050-2},
year = {2018},
date = {2018-04-30},
journal = {Nature Scientific Reports},
volume = {8},
pages = {6750},
abstract = {Carbon nanostructures are promising ballistic protection materials, due to their low density and excellent mechanical properties. Recent experimental and computational investigations on the behavior of graphene under impact conditions revealed exceptional energy absorption properties as well. However, the reported numerical and experimental values differ by an order of magnitude. In this work, we combined numerical and analytical modeling to address this issue. In the numerical part, we employed reactive molecular dynamics to carry out ballistic tests on single, double, and triple-layered graphene sheets. We used velocity values within the range tested in experiments. Our numerical and the experimental results were used to determine parameters for a scaling law. We find that the specific penetration energy decreases as the number of layers (N) increases, from ∼15 MJ/kg for N = 1 to ∼0.9 MJ/kg for N = 350, for an impact velocity of 900 m/s. These values are in good agreement with simulations and experiments, within the entire range of N values for which data is presently available. Scale effects explain the apparent discrepancy between simulations and experiments.},
keywords = {Fracture, Graphene, impact, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
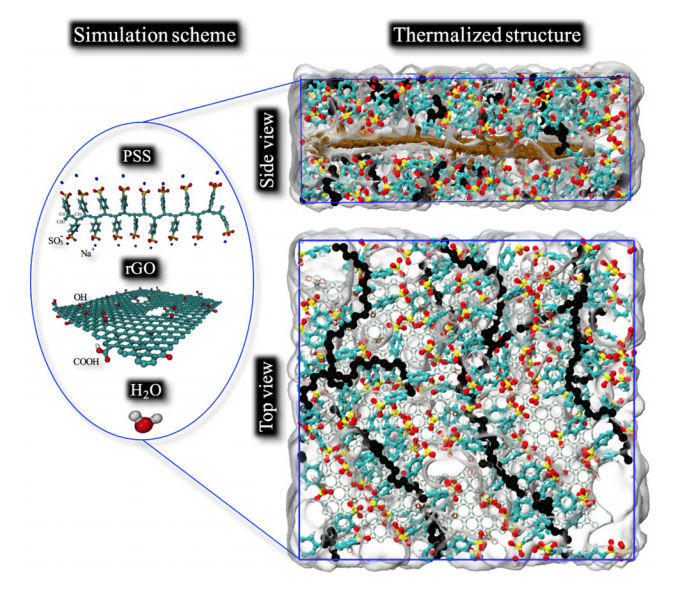
Marco AE Maria Celina M Miyazaki, Daiane Damasceno Borges
Experimental and computational investigation of reduced graphene oxide nanoplatelets stabilized in poly(styrene sulfonate) sodium salt Journal Article
In: Journal of Materials Science, vol. 53, no. 14, pp. 10049-10056, 2018.
Abstract | Links | BibTeX | Tags: Molecular Dynamics, Polymers
@article{Miyazaki2018,
title = {Experimental and computational investigation of reduced graphene oxide nanoplatelets stabilized in poly(styrene sulfonate) sodium salt},
author = {Celina M Miyazaki, Marco AE Maria, Daiane Damasceno Borges, Cristiano F Woellner, Gustavo Brunetto, Alexandre F Fonseca, Carlos JL Constantino, Marcelo A Pereira-da-Silva, Abner de Siervo, Douglas S Galvao, Antonio Riul Jr},
url = {https://link.springer.com/article/10.1007/s10853-018-2325-1},
doi = {https://link.springer.com/article/10.1007/s10853-018-2325-1},
year = {2018},
date = {2018-04-19},
journal = {Journal of Materials Science},
volume = {53},
number = {14},
pages = {10049-10056},
abstract = {The production of large-area interfaces and the use of scalable methods to build up
designed nanostructures generating advanced functional properties are of high
interest for many materials science applications. Nevertheless, large-area coverage
remains a major problem even for pristine graphene, and here we present a hybrid,
composite graphene-like material soluble in water that can be exploited in many
areas such as energy storage, electrodes fabrication, selective membranes and
biosensing. Graphene oxide (GO) was produced by the traditional Hummers’
method being further reduced in the presence of poly(styrene sulfonate) sodium salt
(PSS), thus creating stable reduced graphene oxide (rGO) nanoplatelets wrapped by
PSS (GPSS). Molecular dynamics simulations were carried out to further clarify the
interactions between PSS molecules and rGO nanoplatelets, with calculations
supported by Fourier transform infrared spectroscopy analysis. The intermolecular
forces between rGO nanoplatelets and PSS lead to the formation of a hybrid material
(GPSS) stabilized by van der Waals forces, allowing the fabrication of high-quality
layer-by-layer (LbL) films with poly(allylamine hydrochloride) (PAH). Raman and
electrical characterizations corroborated the successful modifications in the electronic
structures from GO to GPSS after the chemical treatment, resulting in (PAH/
GPSS) LbL films four orders of magnitude more conductive than (PAH/GO).},
keywords = {Molecular Dynamics, Polymers},
pubstate = {published},
tppubtype = {article}
}
designed nanostructures generating advanced functional properties are of high
interest for many materials science applications. Nevertheless, large-area coverage
remains a major problem even for pristine graphene, and here we present a hybrid,
composite graphene-like material soluble in water that can be exploited in many
areas such as energy storage, electrodes fabrication, selective membranes and
biosensing. Graphene oxide (GO) was produced by the traditional Hummers’
method being further reduced in the presence of poly(styrene sulfonate) sodium salt
(PSS), thus creating stable reduced graphene oxide (rGO) nanoplatelets wrapped by
PSS (GPSS). Molecular dynamics simulations were carried out to further clarify the
interactions between PSS molecules and rGO nanoplatelets, with calculations
supported by Fourier transform infrared spectroscopy analysis. The intermolecular
forces between rGO nanoplatelets and PSS lead to the formation of a hybrid material
(GPSS) stabilized by van der Waals forces, allowing the fabrication of high-quality
layer-by-layer (LbL) films with poly(allylamine hydrochloride) (PAH). Raman and
electrical characterizations corroborated the successful modifications in the electronic
structures from GO to GPSS after the chemical treatment, resulting in (PAH/
GPSS) LbL films four orders of magnitude more conductive than (PAH/GO).
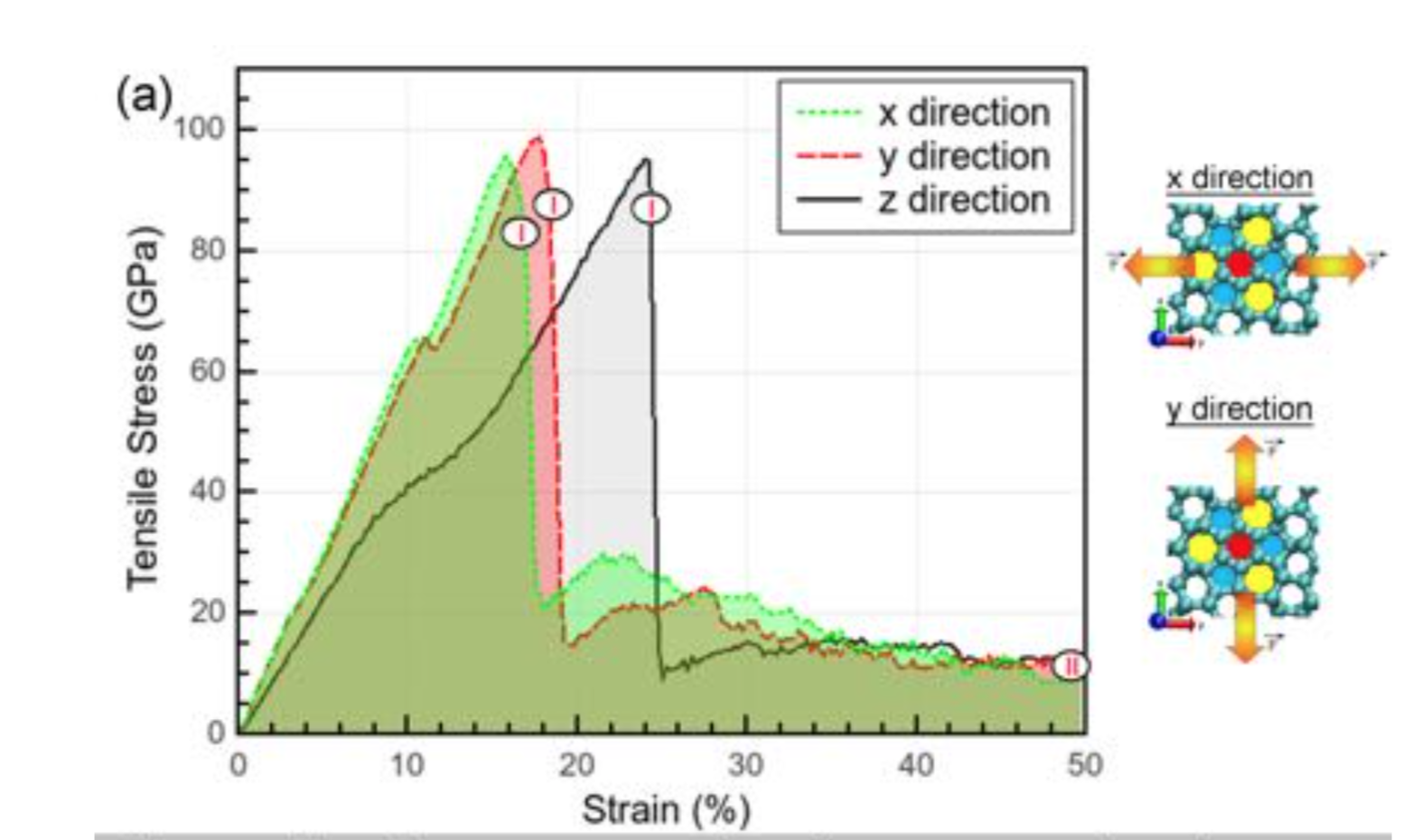
Eliezer F.; Autreto Oliveira, Pedro A. S. ; Woellner
On the mechanical properties of novamene: a fully atomistic molecular dynamics and DFT investigation Online
2018, (preprint ArXiv:1804.07215).
Abstract | Links | BibTeX | Tags: carbon allotropes, DFT, Molecular Dynamics, novamenes
@online{Oliveira2018f,
title = {On the mechanical properties of novamene: a fully atomistic molecular dynamics and DFT investigation},
author = {Oliveira, Eliezer F.; Autreto, Pedro A. S.; Woellner, Cristiano F.; Galvao, Douglas S.},
url = {https://arxiv.org/abs/1804.07215},
year = {2018},
date = {2018-04-19},
abstract = {We have investigated through fully atomistic reactive molecular dynamics and DFT simulations, the mechanical properties and fracture dynamics of novamene, a new 3D carbon allotrope structure recently proposed. Our results showed that novamene is an anisotropic structure with relation to tensile deformation. Although novamente shares some mechanical features with other carbon allotropes, it also exhibits distinct ones, such as, extensive structural reconstructions (self-healing effect). Novamene presents ultimate strength (~ 100 GPa) values lower than other carbon allotropes, but it has the highest ultimate strain along the z-direction (~ 22.5%). Although the Young's modulus (~ 600 GPa) and ultimate strength values are smaller than for other carbon allotropes, they still outperform other materials, such as for example silicon, steel or titanium alloys. With relation to the fracture dynamics, novamene is again anisotropic with the fracture/crack propagation originating from deformed heptagons and pentagons for x and y directions and broken sp3 bonds connecting structural planes. Another interesting feature is the formation of multiple and long carbon linear chains in the final fracture stages.},
note = {preprint ArXiv:1804.07215},
keywords = {carbon allotropes, DFT, Molecular Dynamics, novamenes},
pubstate = {published},
tppubtype = {online}
}
Kabbani, Mohamad A.; Kochat, Vidya; Bhowmick, Sanjit; Soto, Matias; Som, Anirban; Krishnadas, K. R.; Woellner, Cristiano F.; Jaques, Ygor M.; Barrera, Enrique V.; Asif, Syed; Vajtai, Robert; Pradeep, Thalappil; Galvão, Douglas S.; Kabbani, Ahmad T.; Tiwary, Chandra Sekhar; Ajayan, Pulickel M.
Consolidation of Functionalized Graphene at Ambient Temperature via Mechano-chemistry Journal Article
In: Carbon, vol. 134, no. 8, pp. 491-499, 2018.
Abstract | Links | BibTeX | Tags: DFT, Graphene, Mechanochemistry, Molecular Dynamics
@article{Kabbani2018,
title = {Consolidation of Functionalized Graphene at Ambient Temperature via Mechano-chemistry},
author = {Mohamad A. Kabbani and Vidya Kochat and Sanjit Bhowmick and Matias Soto and Anirban Som and K.R. Krishnadas and Cristiano F. Woellner and Ygor M. Jaques and Enrique V. Barrera and Syed Asif and Robert Vajtai and Thalappil Pradeep and Douglas S. Galvão and Ahmad T. Kabbani and Chandra Sekhar Tiwary and Pulickel M. Ajayan},
url = {https://www.sciencedirect.com/science/article/pii/S0008622318302987?dgcid=raven_sd_aip_email},
doi = {DOI:10.1016/j.carbon.2018.03.049},
year = {2018},
date = {2018-03-22},
journal = {Carbon},
volume = {134},
number = {8},
pages = {491-499},
abstract = {Graphitic solids are typically produced via high temperature and energy consuming
processing (e.g. sintering) of carbon particles. Here, we demonstrate the mechano-chemical
assembly of functionalized graphene layers into 3D graphitic solids via room temperature and
low energy consuming processing. The chemical functional groups on graphene layers are
interconnected at room temperature under pressure leading to porous three-dimensional
structures with tunable mechanical and electrical properties. The formation of mechanochemistry
induced atomic scale junctions and their impact on mechanical properties of
graphene assembled carbon materials are demonstrated through nano-indentation experiments
and confirmed using DFT and molecular dynamics simulations. The results show room
temperature consolidation routes of graphene layers into bulk carbon solids.},
keywords = {DFT, Graphene, Mechanochemistry, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
processing (e.g. sintering) of carbon particles. Here, we demonstrate the mechano-chemical
assembly of functionalized graphene layers into 3D graphitic solids via room temperature and
low energy consuming processing. The chemical functional groups on graphene layers are
interconnected at room temperature under pressure leading to porous three-dimensional
structures with tunable mechanical and electrical properties. The formation of mechanochemistry
induced atomic scale junctions and their impact on mechanical properties of
graphene assembled carbon materials are demonstrated through nano-indentation experiments
and confirmed using DFT and molecular dynamics simulations. The results show room
temperature consolidation routes of graphene layers into bulk carbon solids.

Zink, Stefan; Moura, Francisco Alirio; da Silva Autreto, Pedro Alves; Galvao, Douglas Soares; Mizaikoff, Boris
Virtually Imprinted Polymers (VIPs): Understanding Molecularly Templated Materials via Molecular Dynamics Simulations Journal Article
In: Physical Chemistry Chemical Physics, vol. 20, pp. 13145-13152, 2018.
Abstract | Links | BibTeX | Tags: MIPs, Molecular Dynamics
@article{Zink2018b,
title = {Virtually Imprinted Polymers (VIPs): Understanding Molecularly Templated Materials via Molecular Dynamics Simulations},
author = {Stefan Zink and Francisco Alirio Moura and Pedro Alves da Silva Autreto and Douglas Soares Galvao and Boris Mizaikoff},
url = {http://pubs.rsc.org/en/content/articlelanding/2018/cp/c7cp08284c/unauth#!divAbstract},
doi = {10.1039/C7CP08284C},
year = {2018},
date = {2018-02-15},
journal = {Physical Chemistry Chemical Physics},
volume = {20},
pages = {13145-13152},
abstract = {Molecularly imprinted polymers are advanced recognition materials selectively rebinding a target molecule present during synthesis of the polymer matrix. It is commonly understood that the templating process is based on embedding the complex formed between a template and functional monomers into a co-polymer matrix via polymerization with a cross-linker while maintaining their spatial arrangement forming a molecular imprint. Template removal then leads to synthetic recognition sites ready to selectively rebind their targets, which are complementary in functionality, size and shape to the target. In this study, an innovative theoretical concept using fully atomistic molecular dynamics simulations for modeling molecular templating processes is introduced yielding virtually imprinted polymers (VIPs). VIPs created for the template of 17-beta-estradiol and applied in modeled chromatography experiments demonstrated selectivity for their template evidencing the creation of virtual imprints as a result of a template synthesis protocol, which represents a theoretical confirmation of the governing imprinting theory.},
keywords = {MIPs, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Leonardo D Machado Cristiano F Woellner, Pedro AS Autreto; Galvao, Douglas S
Structural Transformations of Carbon and Boron Nitride Nanoscrolls at High Impact Collisions Journal Article
In: Physical Chemistry Chemical Physics, vol. 20, pp. 4911-4916, 2018.
Abstract | Links | BibTeX | Tags: Fracture, impact, Molecular Dynamics, scroll
@article{Woellner2018,
title = {Structural Transformations of Carbon and Boron Nitride Nanoscrolls at High Impact Collisions},
author = {Cristiano F Woellner, Leonardo D Machado, Pedro AS Autreto, Jose M de Sousa, and Douglas S Galvao},
url = {http://pubs.rsc.org/en/content/articlelanding/2018/cp/c7cp07402f#!divAbstract},
doi = {DOI:10.1039/C7CP07402F},
year = {2018},
date = {2018-02-14},
journal = {Physical Chemistry Chemical Physics},
volume = {20},
pages = {4911-4916},
abstract = {The behavior of nanostructures under high strain-rate conditions has been the object of theoretical and
experimental investigations in recent years. For instance, it has been shown that carbon and boron
nitride nanotubes can be unzipped into nanoribbons at high-velocity impacts. However, the response of
many nanostructures to high strain-rate conditions is still unknown. In this work, we have investigated
the mechanical behavior of carbon (CNS) and boron nitride nanoscrolls (BNS) colliding against solid
targets at high velocities, using fully atomistic reactive (ReaxFF) molecular dynamics (MD) simulations.
CNS (BNS) are graphene (boron nitride) membranes rolled up into papyrus-like structures. Their openended
topology leads to unique properties not found in their close-ended analogs, such as nanotubes.
Our results show that collision products are mainly determined by impact velocities and by two
orientation angles, which define the position of the scroll (i) axis and (ii) open edge relative to the target.
Our MD results showed that for appropriate velocities and orientations, large-scale deformations and
nanoscroll fractures could occur. We also observed unscrolling (scrolls going back to quasi-planar
membranes), scroll unzipping into nanoribbons, and significant reconstruction due to breaking and/or
formation of new chemical bonds. For particular edge orientations and velocities, conversion from open
to close-ended topology is also possible, due to the fusion of nanoscroll walls.},
keywords = {Fracture, impact, Molecular Dynamics, scroll},
pubstate = {published},
tppubtype = {article}
}
experimental investigations in recent years. For instance, it has been shown that carbon and boron
nitride nanotubes can be unzipped into nanoribbons at high-velocity impacts. However, the response of
many nanostructures to high strain-rate conditions is still unknown. In this work, we have investigated
the mechanical behavior of carbon (CNS) and boron nitride nanoscrolls (BNS) colliding against solid
targets at high velocities, using fully atomistic reactive (ReaxFF) molecular dynamics (MD) simulations.
CNS (BNS) are graphene (boron nitride) membranes rolled up into papyrus-like structures. Their openended
topology leads to unique properties not found in their close-ended analogs, such as nanotubes.
Our results show that collision products are mainly determined by impact velocities and by two
orientation angles, which define the position of the scroll (i) axis and (ii) open edge relative to the target.
Our MD results showed that for appropriate velocities and orientations, large-scale deformations and
nanoscroll fractures could occur. We also observed unscrolling (scrolls going back to quasi-planar
membranes), scroll unzipping into nanoribbons, and significant reconstruction due to breaking and/or
formation of new chemical bonds. For particular edge orientations and velocities, conversion from open
to close-ended topology is also possible, due to the fusion of nanoscroll walls.
Borges, Daiane Damasceno; Galvao, Douglas S.
Schwarzites for Natural Gas Storage: A Grand-Canonical Monte Carlo Study Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 115-120, 2018.
Abstract | Links | BibTeX | Tags: Gas Storage, Mechanical Properties, Molecular Dynamics, Monte Carlo, Schwarzites
@article{Borges2018d,
title = {Schwarzites for Natural Gas Storage: A Grand-Canonical Monte Carlo Study },
author = {Daiane Damasceno Borges and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/schwarzites-for-natural-gas-storage-a-grandcanonical-monte-carlo-study/2DF8D601AF8EF04BBAC5CCCBEFA8339E},
doi = {https://doi.org/10.1557/adv.2018.190},
year = {2018},
date = {2018-02-13},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = {115-120},
abstract = {he 3D porous carbon-based structures called Schwarzites have been recently a subject of renewed interest due to the possibility of being synthesized in the near future. These structures exhibit negatively curvature topologies with tuneable porous sizes and shapes, which make them natural candidates for applications such as CO2 capture, gas storage and separation. Nevertheless, the adsorption properties of these materials have not been fully investigated. Following this motivation, we have carried out Grand-Canonical Monte Carlo simulations to study the adsorption of small molecules such as CO2, CO, CH4, N2 and H2, in a series of Schwarzites structures. Here, we present our preliminary results on natural gas adsorptive capacity in association with analyses of the guest-host interaction strengths. Our results show that Schwarzites P7par, P8bal and IWPg are the most promising structures with very high CO2 and CH4 adsorption capacity and low saturation pressure (<1bar) at ambient temperature. The P688 is interesting for H2 storage due to its exceptional high H2 adsorption enthalpy value of -19kJ/mol.},
keywords = {Gas Storage, Mechanical Properties, Molecular Dynamics, Monte Carlo, Schwarzites},
pubstate = {published},
tppubtype = {article}
}
Borges, Daiane Damasceno; Woellner, Cristiano F.; Autreto, Pedro A. S.; Galvao, Douglas S.
Water/alcohol separation via layered oxide graphene membranes Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 109-114, 2018.
Abstract | Links | BibTeX | Tags: Filtration, Graphene, Molecular Dynamics
@article{Borges2018d,
title = {Water/alcohol separation via layered oxide graphene membranes},
author = {Daiane Damasceno Borges and Cristiano F. Woellner and Pedro A. S. Autreto and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/wateralcohol-separation-in-graphene-oxide-membranes-insights-from-molecular-dynamics-and-monte-carlo-simulations/C61C66FF48D35EB2DB3408ACCE96C41A},
doi = { https://doi.org/10.1557/adv.2018.192},
year = {2018},
date = {2018-02-13},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = {109-114},
abstract = {Graphene-based membranes have been investigated as promising candidates for water filtration and gas separation applications. Experimental evidences have shown that graphene oxide can be impermeable to liquids, vapors and gases, while allowing a fast permeation of water molecules. This phenomenon has been attributed to the formation of a network of nano capillaries that allow nearly frictionless water flow while blocking other molecules by steric hindrance effects. It is supposed that water molecules are transported through the percolated two-dimensional channels formed between graphene-based sheets. Although these channels allow fast water permeation in such materials, the flow rates are strongly dependent on how the membranes are fabricated. Also, some fundamental issues regarding the nanoscale mechanisms of water permeation are still not fully understood and their interpretation remains controversial. In this work, we have investigated the dynamics of water permeation through pristine graphene and graphene oxide model membranes that have strong impact on water/alcohol separation. We have carried out fully atomistic classical molecular dynamics simulations of systems composed of multiple layered graphene-based sheets into contact with a pure water reservoir under controlled thermodynamics conditions (e. g., by varying temperature and pressure values). We have systematically analysed how the transport dynamics of the confined nanofluids depend on the interlayer distances and the role of the oxide functional groups. Our results show the water flux is much more effective for graphene than for graphene oxide membranes. These results can be attributed to the H-bonds formation between oxide functional groups and water, which traps the water molecules and precludes ultrafast water transport through the nanochannels.},
keywords = {Filtration, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
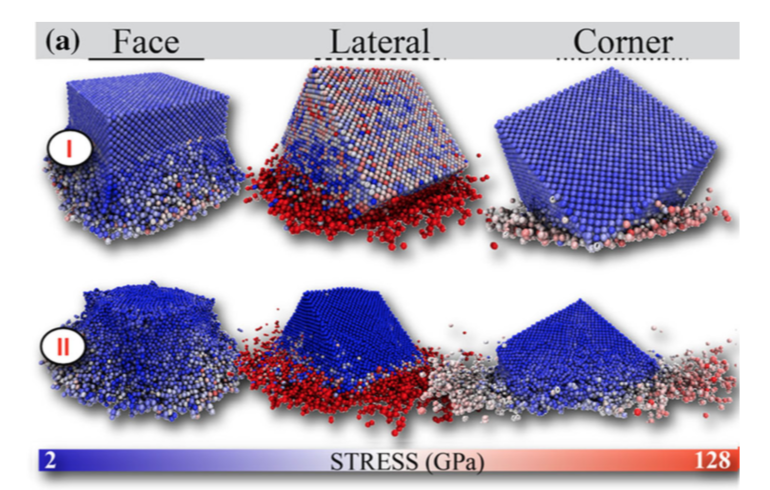
Oliveira, Eliezer Fernando; Autreto, Pedro Alves da Silva; Galvao, Douglas Soares
On hardening silver nanocubes by high velocity impacts: a fully atomistic molecular dynamics investigation Journal Article
In: Journal of Materials Science, vol. 53, no. 10, pp. 7486–7492, 2018.
Abstract | Links | BibTeX | Tags: Fracture, impact, Molecular Dynamics, silver
@article{Oliveira2018,
title = {On hardening silver nanocubes by high velocity impacts: a fully atomistic molecular dynamics investigation},
author = {Oliveira, Eliezer Fernando and Autreto, Pedro Alves da Silva and Galvao, Douglas Soares},
url = {https://link.springer.com/article/10.1007/s10853-018-2104-z},
doi = {10.1007/s10853-018-2104-z},
year = {2018},
date = {2018-02-09},
journal = {Journal of Materials Science},
volume = {53},
number = {10},
pages = {7486–7492},
abstract = {Gradient nanograins (GNG) creation in metals has been a promising approach to obtain ultra-strong materials. Recently, R. Thevamaran et al. (Science 354:312 in 2016) proposed a single-step method based on high-velocity impacts of silver nanocubes (SNC) to produce almost perfect GNG. However, after certain time, these grains spontaneously coalesce, which compromises the induced hardening and other mechanical properties. To better understand these processes, a detailed investigation at the atomic scale of the deformation/hardening mechanisms are needed, which is one of the objectives of the present work. We carried out fully atomistic molecular dynamics (MD) simulations of silver nanocubes at high impact velocity values using realistic structural models. Our MD results suggest that besides the GNG mechanisms, the observed SNC hardening could be also the result of the existence of polycrystalline arrangements formed by HCP domains encapsulated by FCC ones in the smashed SNC. This can be a new way to design ultra-strong materials, even in the absence of GNG domains.},
keywords = {Fracture, impact, Molecular Dynamics, silver},
pubstate = {published},
tppubtype = {article}
}
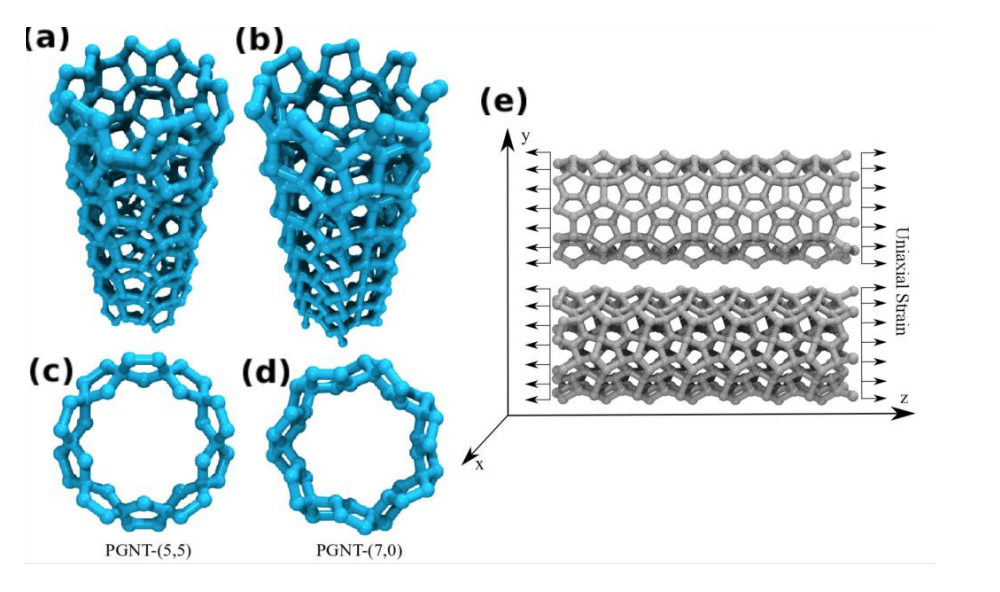
de Sousa, J. M.; Aguiar, A. L.; Girao, E. C.; Fonseca, Alexandre F.; Filho, A. G. Souza; Galvao, Douglas S.
Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 97-102, 2018.
Abstract | Links | BibTeX | Tags: Fracture, Molecular Dynamics, pentagraphene
@article{deSousa2018b,
title = {Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study},
author = {J. M. de Sousa and A. L. Aguiar and E. C. Girao and Alexandre F. Fonseca and A. G. Souza Filho and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-pentagraphenebased-nanotubes-a-molecular-dynamics-study/289AB70DADF20059BB8FCC9EF07B97AB},
doi = { https://doi.org/10.1557/adv.2018.160},
year = {2018},
date = {2018-02-06},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = {97-102},
abstract = {The study of the mechanical properties of nanostructured systems has gained importance in theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of the strongest nanomaterials found in nature, with Young’s Modulus (EY) in the order 1.25 TPa. One interesting question is about the possibility of generating new nanostructures with 1D symmetry and with similar and/or superior CNT properties. In this work, we present a study on the dynamical, structural, mechanical properties, fracture patterns and EY values for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized states) in the same form that CNTs are formed from rolling up graphene membranes. We carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We have considered zigzag-like and armchair-like PGNTs of different diameters. Our results show that PGNTs present EY ∼ 800 GPa with distinct elastic behavior in relation to CNTs, mainly associated with mechanical failure, chirality dependent fracture patterns and extensive structural reconstructions.},
keywords = {Fracture, Molecular Dynamics, pentagraphene},
pubstate = {published},
tppubtype = {article}
}
Cristiano F Woellner Daiane Damasceno Borges, Pedro AS Autreto
Insights on the mechanism of water-alcohol separation in multilayer graphene oxide membranes: entropic versus enthalpic factors Journal Article
In: Carbon, vol. 127, pp. 280-286, 2018.
Abstract | Links | BibTeX | Tags: Filtration, Graphene, Molecular Dynamics
@article{Borges2018,
title = {Insights on the mechanism of water-alcohol separation in multilayer graphene oxide membranes: entropic versus enthalpic factors},
author = {Daiane Damasceno Borges, Cristiano F Woellner, Pedro AS Autreto, Douglas S Galvao},
url = {https://www.sciencedirect.com/science/article/pii/S000862231731134X},
doi = {https://doi.org/10.1016/j.carbon.2017.11.020},
year = {2018},
date = {2018-02-01},
journal = {Carbon},
volume = {127},
pages = {280-286},
abstract = {xperimental evidence has shown that graphene oxide (GO) can be impermeable to liquids, vapors and gases, while it allows a fast permeation of water molecules. Theoretical studies to understand the filtration mechanisms come mostly from water desalination, while very few works have been dedicated to alcohol dehydration. In this work, we have investigated the molecular level mechanism underlying the alcohol/water separation inside GO membranes. A series of Molecular Dynamics and Grand-Canonical Monte Carlo simulations were carried out to probe the ethanol/water and methanol/water separation through GO membranes composed of multiple layered graphene-based films with different interlayer distance values and number of oxygen-containing functional groups. Our results show that the size exclusion and membrane affinities are not sufficient to explain the selectivity. Besides that, the favorable water molecular arrangement inside GO 2D-channels forming a robust H-bond network and the fast water permeation are crucial for an effective separation mechanism. In other words, the separation phenomenon is not only governed by membrane affinities (enthalpic mechanisms) but mainly by the geometry and size factors (entropic mechanisms). Our findings are consistent with the available experimental data and contribute to clarify important aspects of the separation behavior of confined alcohol/water in GO membranes.},
keywords = {Filtration, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
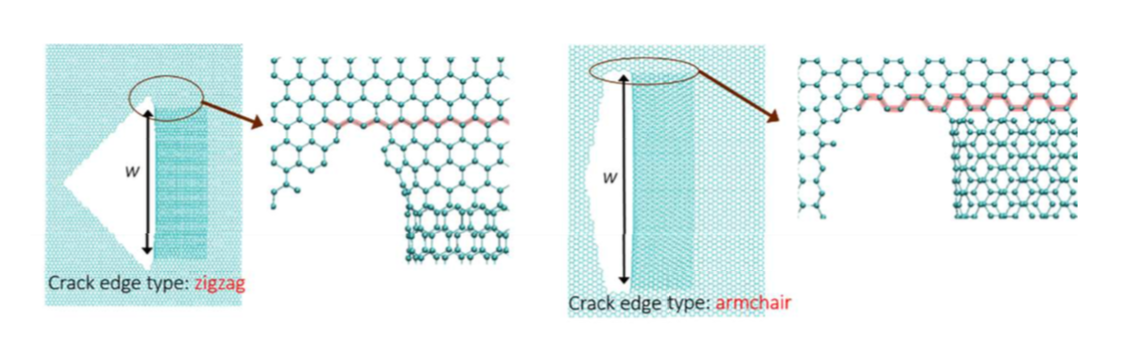
Fonseca, Alexandre F.; Galvao, Douglas S.
Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation Journal Article
In: MRS Advances, vol. 3, no. 8-9, pp. 460-465, 2018.
Abstract | Links | BibTeX | Tags: Fracture, Graphene, Molecular Dynamics
@article{Fonseca2018,
title = {Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation},
author = {Alexandre F. Fonseca and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/selfdriven-graphene-tearing-and-peeling-a-fully-atomistic-molecular-dynamics-investigation/BFC76FC4479AA617E16FA6AC7AB4D487},
doi = {https://doi.org/10.1557/adv.2018.120},
year = {2018},
date = {2018-01-30},
journal = {MRS Advances},
volume = {3},
number = {8-9},
pages = {460-465},
abstract = {In spite of years of intense research, graphene continues to produce surprising results. Recently, it was experimentally observed that under certain conditions graphene can self-drive its tearing and peeling from substrates. This process can generate long, micrometer sized, folded nanoribbons without the action of any external forces. Also, during this cracking-like propagation process, the width of the graphene folded ribbon continuously decreases and the process only stops when the width reaches about few hundreds nanometers in size. It is believed that interplay between the strain energy of folded regions, breaking of carbon-carbon covalent bonds, and adhesion of graphene-graphene and graphene-substrate are the most fundamental features of this process, although the detailed mechanisms at atomic scale remain unclear. In order to gain further insights on these processes we carried out fully atomistic reactive molecular dynamics simulations using the AIREBO potential as available in the LAMMPS computational package. Although the reported tearing/peeling experimental observations were only to micrometer sized structures, our results showed that they could also occur at nanometer scale. Our preliminary results suggest that the graphene tearing/peeling process originates from thermal energy fluctuations that results in broken bonds, followed by strain release that creates a local elastic wave that can either reinforce the process, similar to a whip cracking propagation, or undermine it by producing carbon dangling bonds that evolve to the formation of bonds between the two layers of graphene. As the process continues in time and the folded graphene decreases in width, the carbon-carbon bonds at the ribbon edge and interlayer bonds get less stressed, thermal fluctuations become unable to break them and the process stops.},
keywords = {Fracture, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
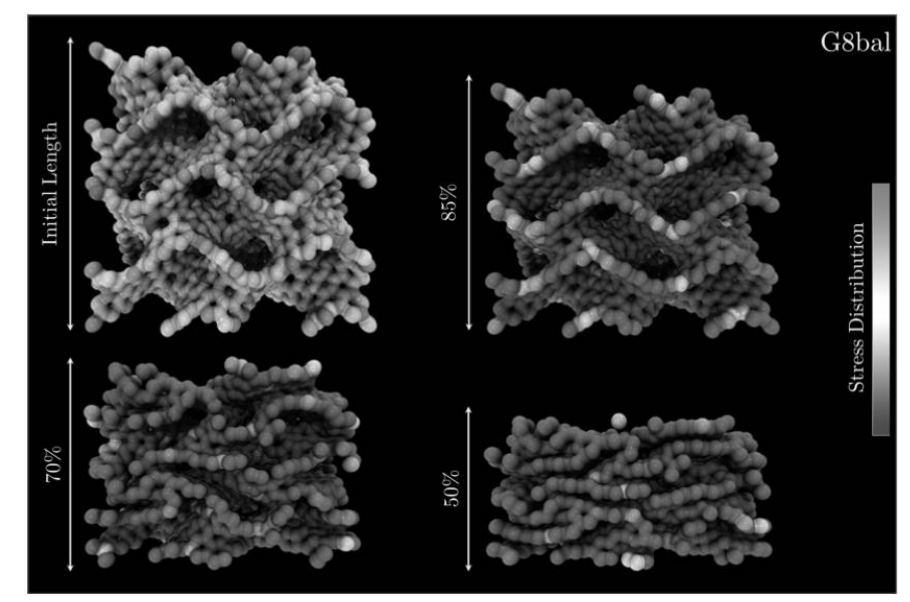
Woellner, Cristiano F.; Botari, Tiago; Perim, Eric; Galvao, Douglas S.
Mechanical Properties of Schwarzites - A Fully Atomistic Reactive Molecular Dynamics Investigation Journal Article
In: MRS Advances, pp. 1-6, 2018.
Abstract | Links | BibTeX | Tags: Mechanical Properties, Molecular Dynamics, Schwarzites
@article{Woellner2018b,
title = {Mechanical Properties of Schwarzites - A Fully Atomistic Reactive Molecular Dynamics Investigation},
author = {Cristiano F. Woellner and Tiago Botari and Eric Perim and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-schwarzites-a-fully-atomistic-reactive-molecular-dynamics-investigation/012AF477491A46541A052C944E4E4834},
doi = { https://doi.org/10.1557/adv.2018.124},
year = {2018},
date = {2018-01-29},
journal = {MRS Advances},
pages = {1-6},
abstract = {Schwarzites are crystalline, 3D porous structures with a stable negative curvature formed of sp2-hybridized carbon atoms. These structures present topologies with tunable porous size and shape and unusual mechanical properties. In this work, we have investigated the mechanical behavior under compressive strain and energy absorption of four different Schwarzites. We considered two Schwarzites families, the so-called Gyroid and Primitive and two structures from each family. We carried out reactive molecular dynamics simulations, using the ReaxFF force field as available in the LAMMPS code. Our results also show they exhibit remarkable resilience under mechanical compression. They can be reduced to half of their original size before structural failure (fracture) occurs.},
keywords = {Mechanical Properties, Molecular Dynamics, Schwarzites},
pubstate = {published},
tppubtype = {article}
}
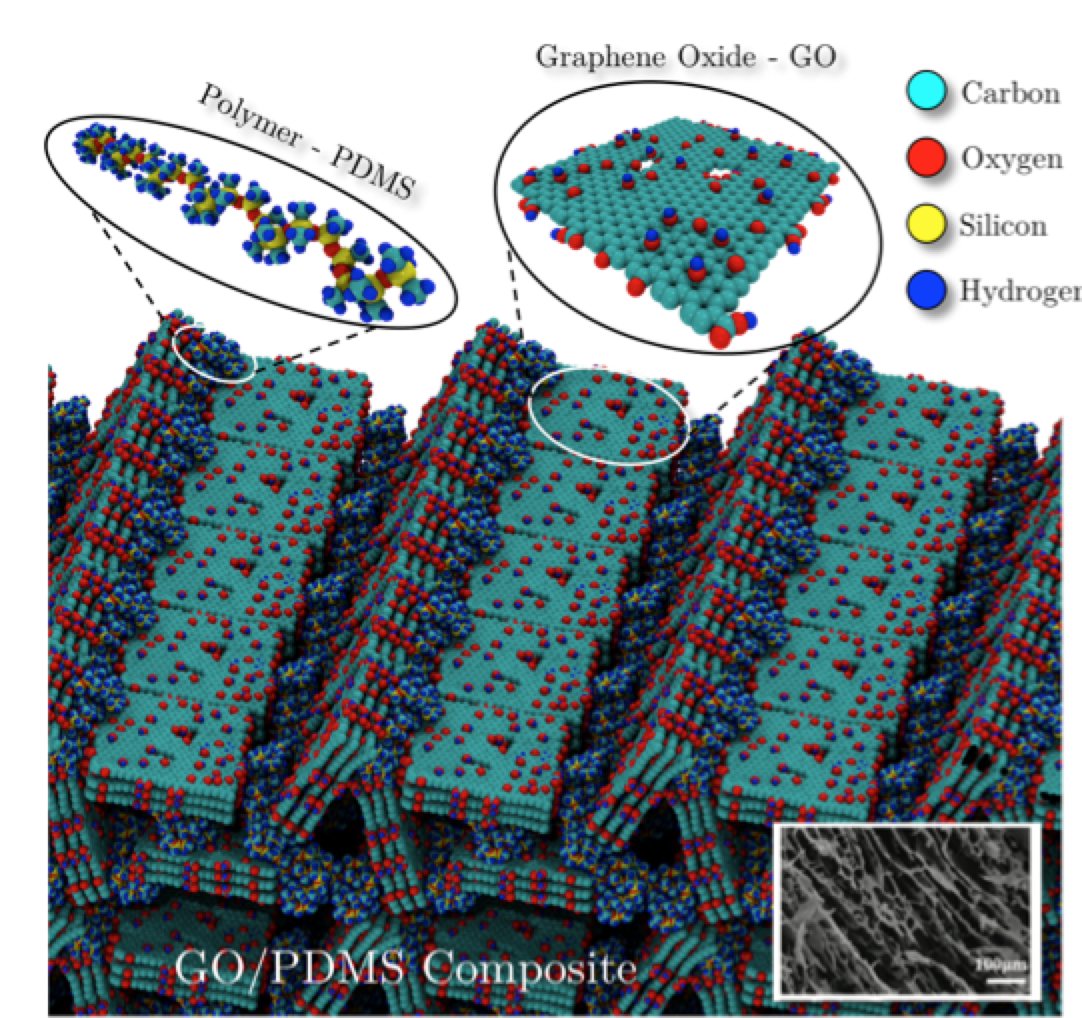
Woellner, Cristiano F.; Owuor, Peter S.; Li, Tong; Vinod, Soumya; Ozden, Sehmus; Kosolwattana, Suppanat; Bhowmick, Sanjit; Duy, Luong X.; Salvatierra, Rodrigo V.; Wei, Bingqing; Asif, Syed A. S.; Tour, James M.; Vajtai, Robert; Lou, Jun; Galvão, Douglas S.; Tiwary, Chandra S.; Ajayan, Pulickel. M.
Mechanical Properties of Ultralow Density Graphene Oxide/Polydimethylsiloxane Foams Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 61-66, 2018.
Abstract | Links | BibTeX | Tags: foams, Mechanical Properties, Molecular Dynamics
@article{Woellner2018c,
title = {Mechanical Properties of Ultralow Density Graphene Oxide/Polydimethylsiloxane Foams},
author = {Cristiano F. Woellner and Peter S. Owuor and Tong Li and Soumya Vinod and Sehmus Ozden and Suppanat Kosolwattana and Sanjit Bhowmick and Luong X. Duy and Rodrigo V. Salvatierra and Bingqing Wei and Syed A. S. Asif and James M. Tour and Robert Vajtai and Jun Lou and Douglas S. Galvão and Chandra S. Tiwary and Pulickel. M. Ajayan},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-ultralow-density-graphene-oxidepolydimethylsiloxane-foams/BC2DC24B3DB5714759FC1EDC71BD9D05},
doi = {DOI: 10.1557/adv.2018. 49},
year = {2018},
date = {2018-01-18},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = { 61-66},
abstract = {Low-density, highly porous graphene/graphene oxide (GO) based-foams have shown high performance in energy absorption applications, even under high compressive deformations. In general, foams are very effective as energy dissipative materials and have been widely used in many areas such as automotive, aerospace and biomedical industries. In the case of graphene-based foams, the good mechanical properties are mainly attributed to the intrinsic graphene and/or GO electronic and mechanical properties. Despite the attractive physical properties of graphene/GO based-foams, their structural and thermal stabilities are still a problem for some applications. For instance, they are easily degraded when placed in flowing solutions, either by the collapsing of their layers or just by structural disintegration into small pieces. Recently, a new and scalable synthetic approach to produce low-density 3D macroscopic GO structure interconnected with polydimethylsiloxane (PDMS) polymeric chains (pGO) was proposed. A controlled amount of PDMS is infused into the freeze-dried foam resulting into a very rigid structure with improved mechanical properties, such as tensile plasticity and toughness. The PDMS wets the graphene oxide sheets and acts like a glue bonding PDMS and GO sheets. In order to obtain further insights on mechanisms behind the enhanced mechanical pGO response we carried out fully atomistic molecular dynamics (MD) simulations. Based on MD results, we build up a structural model that can explain the experimentally observed mechanical behavior.},
keywords = {foams, Mechanical Properties, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
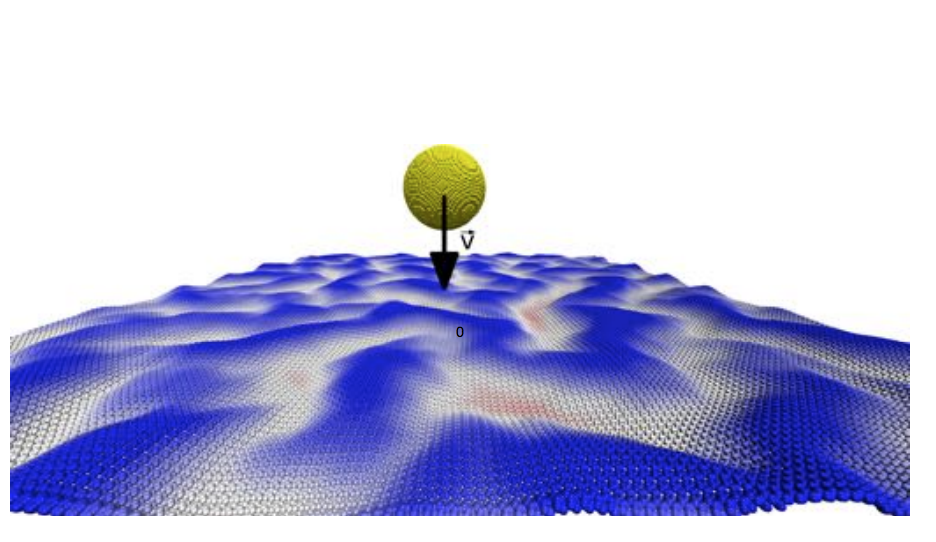
Azevedo, David L.; Bizao, Rafael A.; Galvao, Douglas S.
Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets Online
2018, (preprint arXiv:1801.05346).
Abstract | Links | BibTeX | Tags: Fracture, Molecular Dynamics, pentagraphene
@online{Azevedo2018b,
title = {Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets},
author = {David L. Azevedo and Rafael A. Bizao and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.05346},
year = {2018},
date = {2018-01-18},
abstract = {The superior mechanical properties and low density of carbon nanostructures make them promising ballistic protection materials, stimulating investigations on their high-strain-rate behavior. Recent experiments and simulations revealed graphene possesses exceptional energy absorption properties. In this work, we analyzed through fully atomistic molecular dynamics simulations the ballistic performance of a carbon-based material recently proposed named penta-graphene. Our results show that the fracture pattern is more spherical (no petals formation like observed for graphene). The estimated penetration energy for pentagraphene structures considered here was of 37.69 MJ/Kg, far superior to graphene (29.8 MJ/Kg) under same conditions. These preliminary results are suggestive that pentagraphene could be an excellent material for ballistic applications.},
note = {preprint arXiv:1801.05346},
keywords = {Fracture, Molecular Dynamics, pentagraphene},
pubstate = {published},
tppubtype = {online}
}
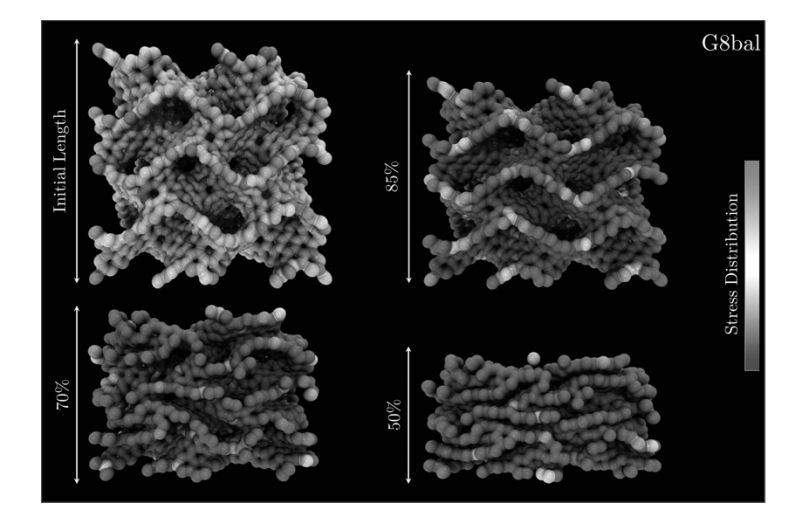
Woellner, Cristiano F.; Botari, Tiago; Perim, Eric; Galvao, Douglas S.
Mechanical Properties of Schwarzites - A Fully Atomistic Reactive Molecular Dynamics Investigation Online
2018, (preprint arXiv:1801.05639).
Abstract | Links | BibTeX | Tags: Mechanical Properties, Molecular Dynamics, Schwarzites
@online{Woellner2018d,
title = {Mechanical Properties of Schwarzites - A Fully Atomistic Reactive Molecular Dynamics Investigation},
author = {Cristiano F. Woellner and Tiago Botari and Eric Perim and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.05639},
year = {2018},
date = {2018-01-18},
abstract = {Schwarzites are crystalline, 3D porous structures with stable negative curvature formed of sp2-hybridized carbon atoms. These structures present topologies with tunable porous size and shape and unusual mechanical properties. In this work, we have investigated the mechanical behavior under compressive strains and energy absorption of four different Schwarzites, through reactive molecular dynamics simulations, using the ReaxFF force field as available in the LAMMPS code. We considered two Schwarzites families, the so-called Gyroid and Primitive and two structures from each family. Our results also show they exhibit remarkable resilience under mechanical compression. They can be reduced to half of their original size before structural failure (fracture) occurs.},
note = {preprint arXiv:1801.05639},
keywords = {Mechanical Properties, Molecular Dynamics, Schwarzites},
pubstate = {published},
tppubtype = {online}
}
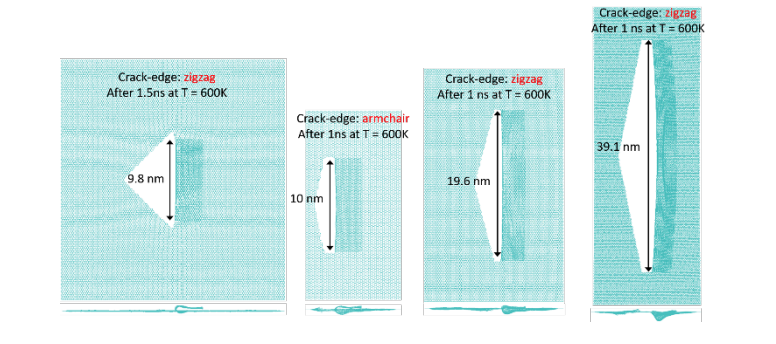
Fonseca, Alexandre F.; Galvao, Douglas S.
Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation Online
2018, (preprint arXiv:1801.05354).
Abstract | Links | BibTeX | Tags: Fracture, Graphene, Molecular Dynamics
@online{Fonseca2018b,
title = {Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation},
author = {Alexandre F. Fonseca and Douglas S. Galvao
},
url = {https://arxiv.org/abs/1801.05354},
year = {2018},
date = {2018-01-17},
abstract = {In spite of years of intense research, graphene continues to produce surprising results. Recently, it was experimentally observed that under certain conditions graphene can self-drive its tearing and peeling from substrates. This process can generate long, micrometer sized, folded nanoribbons without the action of any external forces. Also, during this cracking-like propagation process, the width of the graphene folded ribbon continuously decreases and the process only stops when the width reaches about few hundreds nanometers in size. It is believed that interplay between the strain energy of folded regions, breaking of carbon-carbon covalent bonds, and adhesion of graphene-graphene and graphene-substrate are the most fundamental features of this process, although the detailed mechanisms at atomic scale remain unclear. In order to gain further insights on these processes we carried out fully atomistic reactive molecular dynamics simulations using the AIREBO potential as available in the LAMMPS computational package. Although the reported tearing/peeling experimental observations were only to micrometer sized structures, our results showed that they could also occur at nanometer scale. Our preliminary results suggest that the graphene tearing/peeling process originates from thermal energy fluctuations that results in broken bonds, followed by strain release that creates a local elastic wave that can either reinforce the process, similar to a whip cracking propagation, or undermine it by producing carbon dangling bonds that evolve to the formation of bonds between the two layers of graphene. As the process continues in time and the folded graphene decreases in width, the carbon-carbon bonds at the ribbon edge and interlayer bonds get less stressed, thermal fluctuations become unable to break them and the process stops.},
note = {preprint arXiv:1801.05354},
keywords = {Fracture, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}
de Sousa, J. M.; Aguiar, A. L.; Girao, E. C.; Fonseca, Alexandre F.; Filho, A. G. Souza; Galvao, Douglas S.
Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 67-72, 2018.
Abstract | Links | BibTeX | Tags: Fracture, Molecular Dynamics, phagraphene
@article{deSousa2018c,
title = {Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation},
author = {J. M. de Sousa and A. L. Aguiar and E. C. Girao and Alexandre F. Fonseca and A. G. Souza Filho and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-phagraphene-membranes-a-fully-atomistic-molecular-dynamics-investigation/3ADC3F3B0052AB6632E8681404948E7B},
doi = {DOI: 10.1557/adv.2018. 54},
year = {2018},
date = {2018-01-15},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = {67-72},
abstract = {Recently, a new 2D carbon allotrope structure, named phagraphene (PG), was proposed. PG has a densely array of penta-hexa-hepta-graphene carbon rings. PG was shown to present low and anisotropic thermal conductivity and it is believed that this anisotropy should be also reflected in its mechanical properties. Although PG mechanical properties have been investigated, a detailed and comprehensive study is still lacking. In the present work we have carried out fully atomistic reactive molecular dynamics simulations using the ReaxFF force field, to investigate the mechanical properties and fracture patterns of PG membranes. The Young's modulus values of the PG membranes were estimated from the stress-strain curves. Our results show that these curves present three distinct regimes: one regime where ripples dominate the structure and mechanical properties of the PG membranes; an elastic regime where the membranes exhibit fully planar configurations; and finally am inelastic regime where permanent deformations happened to the PG membrane up to the mechanical failure or fracture.},
keywords = {Fracture, Molecular Dynamics, phagraphene},
pubstate = {published},
tppubtype = {article}
}
de Sousa, J. M.; Aguiar, A. L.; Girao, E. C.; Fonseca, Alexandre F.; Filho, A. G. Sousa; Galvao, Douglas S.
Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study Online
2018, (preprint arXiv:1801.04269).
Abstract | Links | BibTeX | Tags: Fracture, Molecular Dynamics, Nanotubes, pentagraphene
@online{deSousa2018d,
title = {Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study},
author = {J. M. de Sousa and A. L. Aguiar and E. C. Girao and Alexandre F. Fonseca and A. G. Sousa Filho and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.04269},
year = {2018},
date = {2018-01-15},
abstract = {The study of the mechanical properties of nanostructured systems has gained importance in
theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of
the strongest nanomaterials found in nature, with Young's Modulus (YM) in the order 1.25
TPa. One interesting question is about the possibility of generating new nanostructures with
1D symmetry and with similar and/or superior CNT properties. In this work, we present a
study on the dynamical, structural, mechanical properties, fracture patterns and YM values
for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These
tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional
structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized
states) in the same form that CNTs are formed from rolling up graphene membranes. We
carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We
have considered zigzag-like and armchair-like PGNTs of different diameters. Our results
show that PGNTs present YM ~ 800 GPa with distinct elastic behavior in relation to CNTs,
mainly associated with mechanical failure, chirality dependent fracture patterns and extensive
structural reconstructions},
note = {preprint arXiv:1801.04269},
keywords = {Fracture, Molecular Dynamics, Nanotubes, pentagraphene},
pubstate = {published},
tppubtype = {online}
}
theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of
the strongest nanomaterials found in nature, with Young's Modulus (YM) in the order 1.25
TPa. One interesting question is about the possibility of generating new nanostructures with
1D symmetry and with similar and/or superior CNT properties. In this work, we present a
study on the dynamical, structural, mechanical properties, fracture patterns and YM values
for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These
tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional
structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized
states) in the same form that CNTs are formed from rolling up graphene membranes. We
carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We
have considered zigzag-like and armchair-like PGNTs of different diameters. Our results
show that PGNTs present YM ~ 800 GPa with distinct elastic behavior in relation to CNTs,
mainly associated with mechanical failure, chirality dependent fracture patterns and extensive
structural reconstructions
Oliveira, Eliezer Fernando; Santos, Ricardo Paupitz; da Silva Autreto, Pedro Alves; Stanislav Moshkalev,; Galvao, Douglas Soares
Improving Graphene-metal Contacts: Thermal Induced Polishing Online
2018, (preprint ArXiv:1801.04785).
Abstract | Links | BibTeX | Tags: contacts, Graphene, Molecular Dynamics, thermal properties
@online{Oliveira2018d,
title = {Improving Graphene-metal Contacts: Thermal Induced Polishing},
author = {Eliezer Fernando Oliveira and Ricardo Paupitz Santos and Pedro Alves da Silva Autreto and Stanislav Moshkalev, and Douglas Soares Galvao},
url = {https://arxiv.org/abs/1801.04785},
year = {2018},
date = {2018-01-15},
abstract = {Graphene is a very promising material for nanoelectronics applications due to its unique and remarkable electronic and thermal properties. However, when deposited on metallic electrodes the overall thermal conductivity is significantly decreased. This phenomenon has been attributed to the mismatch between the interfaces and contact thermal resistance. Experimentally, one way to improve the graphene/metal contact is thorough high-temperature annealing, but the detailed mechanisms behind these processes remain unclear. In order to address these questions, we carried out fully atomistic reactive molecular dynamics simulations using the ReaxFF force field to investigate the interactions between multi-layer graphene and metallic electrodes (nickel) under (thermal) annealing. Our results show that the annealing induces an upward-downward movement of the graphene layers, causing a pile- driver-like effect over the metallic surface. This graphene induced movements cause a planarization (thermal polishing-like effect) of the metallic surface, which results in the increase of the effective graphene/metal contact area. This can also explain the experimentally observed improvements of the thermal and electric conductivities.},
note = {preprint ArXiv:1801.04785},
keywords = {contacts, Graphene, Molecular Dynamics, thermal properties},
pubstate = {published},
tppubtype = {online}
}

de Sousa, Jose M.; Aguiar, Acrisio L.; Girao, Eduardo C.; Fonseca, Alexandre F.; Antonio G. Sousa Filho,; Galvao, Douglas S.
Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation Online
2018, (preprint arXiv:1801.04292).
Abstract | Links | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, phagraphene
@online{deSousa2018e,
title = {Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Jose M. de Sousa and Acrisio L. Aguiar and Eduardo C. Girao and Alexandre F. Fonseca and Antonio G. Sousa Filho, and Douglas S. Galvao
},
url = {https://arxiv.org/abs/1801.04292},
year = {2018},
date = {2018-01-12},
abstract = {Recently, a new 2D carbon allotrope structure, named phagraphene (PG), was proposed. PG has a densely array of penta-hexa-hepta-graphene carbon rings. PG was shown to present low and anisotropic thermal conductivity and it is believed that this anisotropy should be also reflected in its mechanical properties. Although PG mechanical properties have been investigated, a detailed and comprehensive study is still lacking. In the present work we have carried out fully atomistic reactive molecular dynamics simulations using the ReaxFF force field, to investigate the mechanical properties and fracture patterns of PG membranes. The Young's modulus values of the PG membranes were estimated from the stress-strain curves. Our results show that these curves present three distinct regimes: one regime where ripples dominate the structure and mechanical properties of the PG membranes; an elastic regime where the membranes exhibit fully planar configurations; and finally a plastic regime where permanent deformations happened to the PG membrane up to the mechanical failure or fracture.},
note = {preprint arXiv:1801.04292},
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, phagraphene},
pubstate = {published},
tppubtype = {online}
}
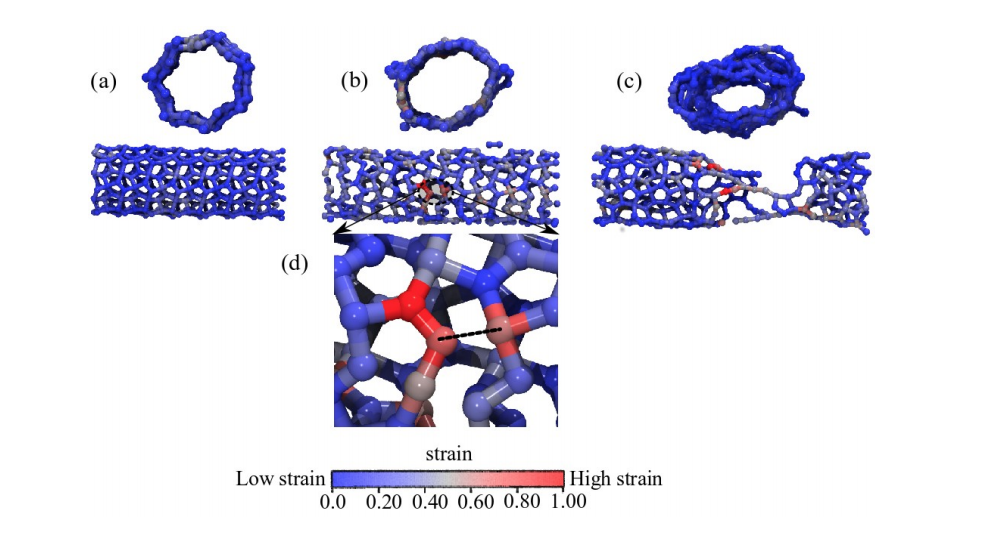
de Sousa, Jose M.; Aguiar, Acrisio L.; Girao, Eduardo C.; Fonseca, Alexandre F.; Antonio G. Souza Filho,; Galvao, Douglas S.
Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study Online
2018, (preprint arXiv:1801.04269).
Abstract | Links | BibTeX | Tags: Fracture, Molecular Dynamics, pentagraphene
@online{deSousa2018f,
title = {Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study},
author = {Jose M. de Sousa and Acrisio L. Aguiar and Eduardo C. Girao and Alexandre F. Fonseca and Antonio G. Souza Filho, and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.04269},
year = {2018},
date = {2018-01-12},
abstract = {The study of the mechanical properties of nanostructured systems has gained importance in theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of the strongest nanomaterials found in nature, with Young's Modulus (YM) in the order 1.25 TPa. One interesting question is about the possibility of generating new nanostructures with 1D symmetry and with similar and/or superior CNT properties. In this work, we present a study on the dynamical, structural, mechanical properties, fracture patterns and YM values for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized states) in the same form that CNTs are formed from rolling up graphene membranes. We carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We have considered zigzag-like and armchair-like PGNTs of different diameters. Our results show that PGNTs present YM ~ 800 GPa with distinct elastic behavior in relation to CNTs, mainly associated with mechanical failure, chirality dependent fracture patterns and extensive structural reconstructions.},
note = {preprint arXiv:1801.04269},
keywords = {Fracture, Molecular Dynamics, pentagraphene},
pubstate = {published},
tppubtype = {online}
}

Azevedo, David L.; Bizao, Rafael A.; Galvao, Douglas S.
Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets Journal Article
In: MRS Advances, vol. 3, no. 8-9, pp. 431-435, 2018.
Abstract | Links | BibTeX | Tags: Fracture, Molecular Dynamics, pentagraphene
@article{Azevedo2018,
title = {Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets},
author = {David L. Azevedo and Rafael A. Bizao and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/molecular-dynamics-simulations-of-ballistic-penetration-of-pentagraphene-sheets/8759C0815840EDE83896EF4A17278228},
doi = {https://doi.org/10.1557/adv.2018.61},
year = {2018},
date = {2018-01-06},
journal = {MRS Advances},
volume = {3},
number = {8-9},
pages = {431-435},
abstract = {The search for new materials with low density and superior mechanical properties is a very intense and stimulating investigation area. These new materials could provide potential application for ballistic protection. Recent experiments and simulations revealed graphene possesses exceptional energy absorption properties. In this work, we analysed through fully atomistic molecular dynamics simulations the ballistic performance of a carbon-based material recently proposed named penta-graphene. Our results show that the fracture pattern is more spherical (no petals formation like observed for graphene). The estimated penetration energy for single-layer penta-graphene structures obtained here was d_1penta∼37.7 MJ/kg, and is comparable with recently results obtained for graphene: d_(1graphene)∼29.0 MJ/kg and d_(1graphene)∼40.8 MJ/kg under similar conditions. These preliminary results are suggestive that penta-graphene could be an excellent material for ballistic applications.},
keywords = {Fracture, Molecular Dynamics, pentagraphene},
pubstate = {published},
tppubtype = {article}
}

M, Ajayan Pulickel; Woellner, Cristiano F; Owuor, Peter S; Trigueiro, Joao P C; Machado, Leonardo D; Silva, Wellington M; Kosolwattana, Suppanat; Jaques, Ygor M; Silva, Carlos J R; Pedrotti, Jairo; Tiwary, Chandra S; Chipara, Alin C; Galvao, Douglas; Chopra, Nitin; Odeh, Ihab N; Silva, Glaura G.
Hybrid 2D Nanostructures for Mechanical Reinforcement and Thermal Conductivity Enhancement in Polymer Composite Journal Article
In: Composites Science and Technology, vol. 159, no. 5, pp. 103-110, 2018.
Abstract | Links | BibTeX | Tags: Composites, Molecular Dynamics
@article{M2018,
title = {Hybrid 2D Nanostructures for Mechanical Reinforcement and Thermal Conductivity Enhancement in Polymer Composite},
author = {Ajayan Pulickel M and Cristiano F Woellner and Peter S Owuor and Joao P C Trigueiro and Leonardo D Machado and Wellington M Silva and Suppanat Kosolwattana and Ygor M Jaques and Carlos J R Silva and Jairo Pedrotti and Chandra S Tiwary and Alin C Chipara and Douglas Galvao and Nitin Chopra and Ihab N Odeh and Glaura G. Silva
},
doi = {https://doi.org/10.1016/j.compscitech.2018.01.032},
year = {2018},
date = {2018-01-01},
journal = {Composites Science and Technology},
volume = {159},
number = {5},
pages = {103-110},
abstract = {Hexagonal boron nitride (h-BN), graphene oxide (GO) and hybrid (GO/h-BN) nanosheets were employed as fillers in order to enhance the physical properties of the polymer matrix. Composites based in epoxy and these two-dimensional (2D) nanofillers were produced with different wt% and their microstructure, mechanical and thermal properties were investigated. Increases up to 140% in tensile strength, 177% in ultimate strain and 32% in elastic modulus were observed for the hybrid GO/h-BN composite with 0.5 wt% content. The hybrid nanofiller also contributed to the increase up to 142% on thermal conductivity with respect to the pure epoxy for GO/h-BN composite with 2.0 wt% content. Molecular dynamic simulation was used to predict the behavior of possible stacking arrangements between h-BN and GO nanosheets tensioned by normal and shear forces. The results showed that the hybrid GO/h-BN combination can prevent the re-stacking process of exfoliated layers, demonstrating the synergism between these nanostructures with the final effect of better dispersion in the composite material. The excellent thermal and mechanical performance of these hybrid composites en- gineered by the combination of different types of the 2D inorganic nanoparticles make them multifunctional candidates for advanced materials applications.},
keywords = {Composites, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
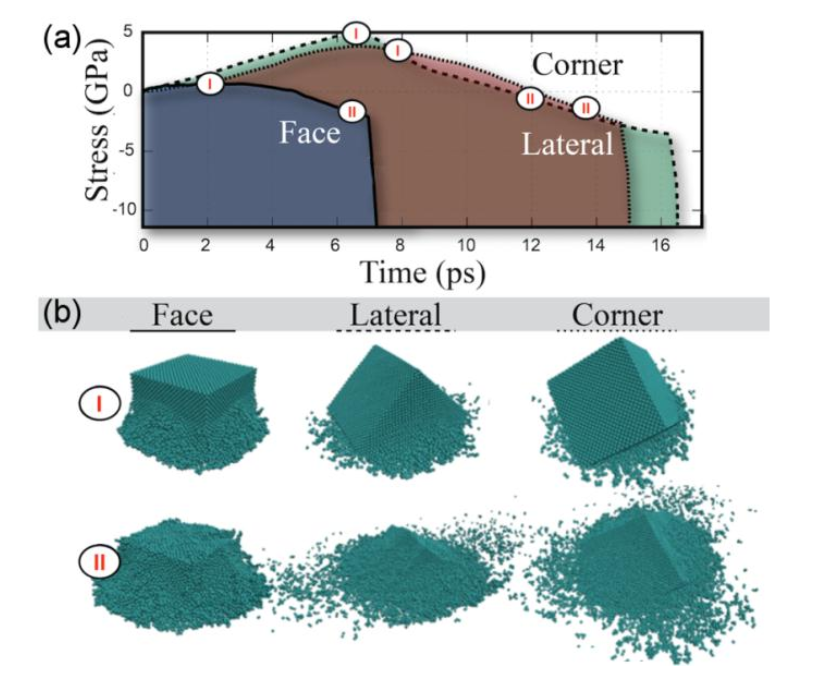
Oliveira, Eliezer Fernando; da Silva Autreto, Pedro Alves; Galvao, Douglas Soares
Silver Hardening via Hypersonic Impacts Journal Article
In: MRS Advances, vol. 3, no. 8-9, pp. 489-494, 2018.
Abstract | Links | BibTeX | Tags: Fracture, impact, Molecular Dynamics, silver
@article{Oliveira2018b,
title = {Silver Hardening via Hypersonic Impacts},
author = {Eliezer Fernando Oliveira and Pedro Alves da Silva Autreto and Douglas Soares Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/silver-hardening-via-hypersonic-impacts/6A35FAB117B4FD244BBD11A64CD25160},
doi = {DOI: 10.1557/adv.2018. 173},
year = {2018},
date = {2018-01-01},
journal = {MRS Advances},
volume = {3},
number = {8-9},
pages = {489-494},
abstract = {The search for new ultra strong materials has been a very active research area. With relation to metals, a successful way to improve their strength is by the creation of a gradient of nanograins (GNG) inside the material. Recently, R. Thevamaran et al. [Science v354, 312- 316 (2016)] propose a single step method based on high velocity impact of silver nanocubes to produce high-quality GNG. This method consists of producing high impact collisions of silver cubes at hypersonic velocity (~400 m/s) against a rigid wall. Although they observed an improvement in the mechanical properties of the silver after the impact, the GNG creation and the strengthening mechanism at nanoscale remain unclear. In order to gain further insights about these mechanisms, we carried out fully atomistic molecular dynamics simulations (MD) to investigate the atomic conformations/rearrangements during and after high impact collisions of silver nanocubes at ultrasonic velocity. Our results indicate the co- existence of polycrystalline arrangements after the impact formed by core HCP domains surrounded by FCC ones, which could also contribute to explain the structural hardening.},
keywords = {Fracture, impact, Molecular Dynamics, silver},
pubstate = {published},
tppubtype = {article}
}
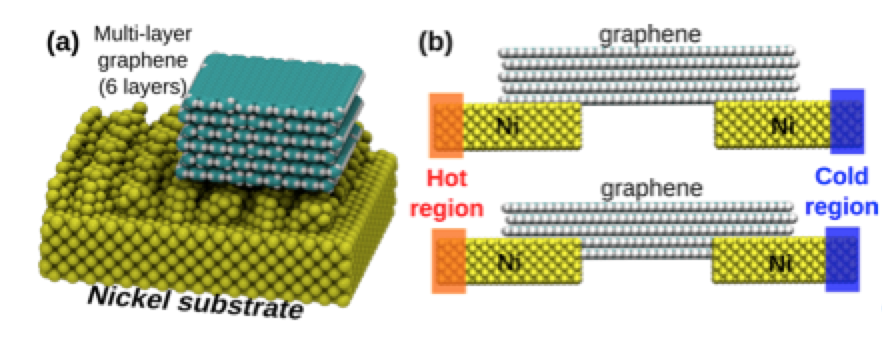
Oliveira, Eliezer Fernando; Paupitz, Ricardo; da Silva Autreto, Pedro Alves; Moshkalev, Stanislav; Galvao, Douglas Soares
Improving Graphene-metal Contacts: Thermal Induced Polishing Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 73-78, 2018.
Abstract | Links | BibTeX | Tags: contacts, Graphene, Molecular Dynamics, thermal properties
@article{Oliveira2018c,
title = {Improving Graphene-metal Contacts: Thermal Induced Polishing },
author = {Eliezer Fernando Oliveira and Ricardo Paupitz and Pedro Alves da Silva Autreto and Stanislav Moshkalev and Douglas Soares Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/improving-graphenemetal-contacts-thermal-induced-polishing/AC01C4996B90B0EE5E03220604071D12},
doi = {https://doi.org/10.1557/adv.2018.66},
year = {2018},
date = {2018-01-01},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = {73-78},
abstract = {Graphene is a very promising material for nanoelectronics applications due to its unique and remarkable electronic and thermal properties. However, when deposited on metallic electrodes the overall thermal conductivity is significantly decreased. This phenomenon has been attributed to the mismatch between the interfaces and contact thermal resistance. Experimentally, one way to improve the graphene/metal contact is thorough high-temperature annealing, but the detailed mechanisms behind these processes remain unclear. In order to address these questions, we carried out fully atomistic reactive molecular dynamics simulations using the ReaxFF force field to investigate the interactions between multi-layer graphene and metallic electrodes (nickel) under (thermal) annealing. Our results show that the annealing induces an upward-downward movement of the graphene layers, causing a pile-driver-like effect over the metallic surface. This graphene induced movements cause a planarization (thermal polishing-like effect) of the metallic surface, which results in the increase of the effective graphene/metal contact area. This can also explain the experimentally observed improvements of the thermal and electric conductivities.},
keywords = {contacts, Graphene, Molecular Dynamics, thermal properties},
pubstate = {published},
tppubtype = {article}
}
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ

