
Autreto, Pedro A. S.; Galvao, Douglas S.
Site Dependent Hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation Journal Article
In: Mater. Res. Soc. Symp. Proc. , vol. 1726 , 2015.
@article{Autreto2015,
title = {Site Dependent Hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Pedro A. S. Autreto and Douglas S. Galvao},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=9702693&fulltextType=RA&fileId=S1946427415004649},
doi = {10.1557/opl.2015.464},
year = {2015},
date = {2015-05-22},
journal = {Mater. Res. Soc. Symp. Proc. },
volume = {1726 },
abstract = {Graphyne is a generic name for a carbon allotrope family of 2D structures, where acetylenic groups connect benzenoid rings, with the coexistence of sp and sp2 hybridized carbon atoms. In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics and structural changes of the hydrogenation of α, β, and γ graphyne forms. Our results showed that the existence of different sites for hydrogen bonding, related to single and triple bonds, makes the process of incorporating hydrogen atoms into graphyne membranes much more complex than the graphene ones. Our results also show that hydrogenation reactions are strongly site dependent and that the sp-hybridized carbon atoms are the preferential sites to chemical attacks. In our cases, the effectiveness of the hydrogenation (estimated from the number of hydrogen atoms covalently bonded to carbon atoms) follows the α, β, γ-graphyne structure ordering.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Perim Martins, Eric; Paupitz, Ricardo; Autreto, Pedro Alves da Silva; Galvao, Douglas Soares
Inorganic Graphenylene: A Porous Two-Dimensional Material with Tunable Band Gap Journal Article
In: The Journal of Physical Chemistry C, vol. 118, no. 41, pp. 23670–23674, 2014.
@article{perim2014inorganic,
title = {Inorganic Graphenylene: A Porous Two-Dimensional Material with Tunable Band Gap},
author = {Perim Martins, Eric and Paupitz, Ricardo and Autreto, Pedro Alves da Silva and Galvao, Douglas Soares},
url = {http://pubs.acs.org/doi/abs/10.1021/jp502119y},
year = {2014},
date = {2014-01-01},
journal = {The Journal of Physical Chemistry C},
volume = {118},
number = {41},
pages = {23670–23674},
publisher = {American Chemical Society},
abstract = {By means of ab initio calculations, we investigate the possibility of existence of a boron nitride (BN) porous two-dimensional nanosheet, which is geometrically similar to the carbon allotrope known as biphenylene carbon. The proposed structure, which we called inorganic graphenylene (IGP), is formed spontaneously after selective dehydrogenation of the porous boron nitride (BN) structure proposed by Ding et al. We study the structural and electronic properties of both porous BN and IGP, and it is shown that, by selective substitution of B and N atoms with carbon atoms in these structures, the band gap can be significantly reduced, changing their behavior from insulators to semiconductors, thus opening the possibility of band gap engineering for this class of two-dimensional materials.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
E. Perim T. Botari, P. A. S. Autreto
Mechanical properties and fracture dynamics of silicene membranes Journal Article
In: Phys. Chem. Chem. Phys., vol. 16, pp. 19417–19423, 2014.
@article{t2014mechanical,
title = {Mechanical properties and fracture dynamics of silicene membranes},
author = {T. Botari, E. Perim, P. A. S. Autreto, A. C. T. van Duin, R. Paupitz, D. S. Galvao},
url = {http://pubs.rsc.org/en/Content/ArticleLanding/2014/CP/C4CP02902J#!divAbstract},
year = {2014},
date = {2014-01-01},
journal = {Phys. Chem. Chem. Phys.},
volume = {16},
pages = {19417--19423},
abstract = {As graphene has become one of the most important materials, there is renewed interest in other similar structures. One example is silicene, the silicon analogue of graphene. It shares some of the remarkable graphene properties, such as the Dirac cone, but presents some distinct ones, such as a pronounced structural buckling. We have investigated, through density functional based tight-binding (DFTB), as well as reactive molecular dynamics (using ReaxFF), the mechanical properties of suspended single-layer silicene. We calculated the elastic constants, analyzed the fracture patterns and edge reconstructions. We also addressed the stress distributions, unbuckling mechanisms and the fracture dependence on the temperature. We analysed the differences due to distinct edge morphologies, namely zigzag and armchair.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Autreto, PAS; de Sousa, JM; Galvao, DS
Site-dependent hydrogenation on graphdiyne Journal Article
In: Carbon, vol. 77, pp. 829–834, 2014.
@article{Autreto2014,
title = {Site-dependent hydrogenation on graphdiyne},
author = {Autreto, PAS and de Sousa, JM and Galvao, DS},
url = {http://www.sciencedirect.com/science/article/pii/S0008622314005429},
year = {2014},
date = {2014-01-01},
journal = {Carbon},
volume = {77},
pages = {829--834},
publisher = {Pergamon},
abstract = {Graphene is one of the most important materials in science today due to its unique and remarkable electronic, thermal and mechanical properties. However in its pristine state, graphene is a gapless semiconductor, what limits its use in transistor electronics. In part due to the revolution created by graphene in materials science, there is a renewed interest in other possible graphene-like two-dimensional structures. Examples of these structures are graphynes and graphdiynes, which are two-dimensional structures, composed of carbon atoms in sp2 and sp-hybridized states. Graphdiynes (benzenoid rings connecting two acetylenic groups) were recently synthesized and some of them are intrinsically nonzero gap systems. These systems can be easily hydrogenated and the relative level of hydrogenation can be used to tune the band gap values. We have investigated, using fully reactive molecular dynamics (ReaxFF), the structural and dynamics aspects of the hydrogenation mechanisms of graphdiyne membranes. Our results showed that the hydrogen bindings have different atom incorporation rates and that the hydrogenation patterns change in time in a very complex way. The formation of correlated domains reported to hydrogenated graphene is no longer observed in graphdiyne cases.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Vinod, Soumya; Tiwary, Chandra Sekhar; da Silva Autreto, Pedro Alves; Taha-Tijerina, Jaime; Ozden, Sehmus; Chipara, Alin Cristian; Vajtai, Robert; Galvao, Douglas S; Narayanan, Tharangattu N; Ajayan, Pulickel M
Low-density three-dimensional foam using self-reinforced hybrid two-dimensional atomic layers Journal Article
In: Nature communications, vol. 5, 2014.
@article{Vinod2014,
title = {Low-density three-dimensional foam using self-reinforced hybrid two-dimensional atomic layers},
author = {Vinod, Soumya and Tiwary, Chandra Sekhar and da Silva Autreto, Pedro Alves and Taha-Tijerina, Jaime and Ozden, Sehmus and Chipara, Alin Cristian and Vajtai, Robert and Galvao, Douglas S and Narayanan, Tharangattu N and Ajayan, Pulickel M},
url = {http://www.nature.com/ncomms/2014/140729/ncomms5541/full/ncomms5541.html},
year = {2014},
date = {2014-01-01},
journal = {Nature communications},
volume = {5},
publisher = {Nature Publishing Group},
abstract = {Low-density nanostructured foams are often limited in applications due to their low mechanical and thermal stabilities. Here we report an approach of building the structural units of three-dimensional (3D) foams using hybrid two-dimensional (2D) atomic layers made of stacked graphene oxide layers reinforced with conformal hexagonal boron nitride (h-BN) platelets. The ultra-low density (1/400 times density of graphite) 3D porous structures are scalably synthesized using solution processing method. A layered 3D foam structure forms due to presence of h-BN and significant improvements in the mechanical properties are observed for the hybrid foam structures, over a range of temperatures, compared with pristine graphene oxide or reduced graphene oxide foams. It is found that domains of h-BN layers on the graphene oxide framework help to reinforce the 2D structural units, providing the observed improvement in mechanical integrity of the 3D foam structure.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Perim, E; Autreto, PAS; Paupitz, R; Galvao, DS
Dynamical aspects of the unzipping of multiwalled boron nitride nanotubes Journal Article
In: Physical Chemistry Chemical Physics, vol. 15, no. 44, pp. 19147–19150, 2013.
@article{perim2013dynamical,
title = {Dynamical aspects of the unzipping of multiwalled boron nitride nanotubes},
author = {Perim, E and Autreto, PAS and Paupitz, R and Galvao, DS},
year = {2013},
date = {2013-01-01},
journal = {Physical Chemistry Chemical Physics},
volume = {15},
number = {44},
pages = {19147--19150},
publisher = {Royal Society of Chemistry},
abstract = {Boron nitride nanoribbons (BNNRs) exhibit very interesting magnetic properties, which could be very useful in the development of spintronic based devices. One possible route to obtain BNNRs is through the unzipping of boron nitride nanotubes (BNNTs), which have been already experimentally realized. In this work, different aspects of the unzipping process of BNNTs were investigated through fully atomistic molecular dynamics simulations using a classical reactive force field (ReaxFF). We investigated multiwalled BNNTs of different diameters and chiralities. Our results show that chirality plays a very important role in the unzipping process, as well as the interlayer coupling. These combined aspects significantly change the fracturing patterns and several other features of the unzipping processes in comparison to the ones observed for carbon nanotubes. Also, similar to carbon nanotubes, defective BNNTs can create regions of very high curvature which can act as a path to the unzipping process.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Paupitz, R; Autreto, Pedro AS; Legoas, SB; Srinivasan, S Goverapet; van Duin, Adri CT; Galvao, DS
Graphene to fluorographene and fluorographane: a theoretical study Journal Article
In: Nanotechnology, vol. 24, no. 3, pp. 035706, 2013.
@article{Paupitz2013,
title = {Graphene to fluorographene and fluorographane: a theoretical study},
author = {Paupitz, R and Autreto, Pedro AS and Legoas, SB and Srinivasan, S Goverapet and van Duin, Adri CT and Galvao, DS},
url = {http://iopscience.iop.org/0957-4484/24/3/035706/article?fromSearchPage=true},
year = {2013},
date = {2013-01-01},
journal = {Nanotechnology},
volume = {24},
number = {3},
pages = {035706},
publisher = {IOP Publishing},
abstract = {We report here a fully reactive molecular dynamics study on the structural and dynamical aspects of the fluorination of graphene membranes (fluorographene). Our results show that fluorination tends to produce defective areas on the graphene membranes with significant distortions of carbon–carbon bonds. Depending on the amount of incorporated fluorine atoms, large membrane holes were observed due to carbon atom losses. These results may explain the broad distribution of the structural lattice parameter values experimentally observed. We have also investigated the effects of mixing hydrogen and fluorine atoms on the graphene functionalization. Our results show that, when in small amounts, the presence of hydrogen atoms produces a significant decrease in the rate of fluorine incorporation onto the membrane. On the other hand, when fluorine is the minority element, it produces a significant catalytic effect on the rate of hydrogen incorporation. We have also observed the spontaneous formation of new hybrid structures with different stable configurations (chair-like, zigzag-like and boat-like) which we named fluorographane.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Perim, Eric; Santos, Ricardo Paupitz; Autreto, Pedro Alves da Silva; Galvao, Douglas S
Fracture Patterns of Boron Nitride Nanotubes Journal Article
In: MRS Proceedings, vol. 1526, pp. mrsf12–1526, 2013.
@article{Perim2013,
title = {Fracture Patterns of Boron Nitride Nanotubes},
author = {Perim, Eric and Santos, Ricardo Paupitz and Autreto, Pedro Alves da Silva and Galvao, Douglas S},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1526},
pages = {mrsf12--1526},
publisher = {Cambridge University Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Machado, LD; Autreto, PAS; Galvao, DS
Graphyne Oxidation: Insights From a Reactive Molecular Dynamics Investigation Journal Article
In: MRS Proceedings, vol. 1549, pp. 53–58, 2013.
@article{Machado2013,
title = {Graphyne Oxidation: Insights From a Reactive Molecular Dynamics Investigation},
author = {Machado, LD and Autreto, PAS and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8963025&fileId=S194642741300941X},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1549},
pages = {53--58},
publisher = {Cambridge University Press},
abstract = {Graphyne is a generic name for a family of carbon allotrope two-dimensional structures where sp2 (single and double bonds) and sp (triple bonds) hybridized states coexists. They exhibit very interesting electronic and mechanical properties sharing some of the unique graphene characteristics. Similarly to graphene, the graphyne electronic properties can be modified by chemical functionalization, such as; hydrogenation, fluorination and oxidation. Oxidation is of particular interest since it can produce significant structural damages.
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics and structural changes of the oxidation of single-layer graphyne membranes at room temperature. We have considered α, β, and γ-graphyne structures. Our results showed that the oxidation reactions are strongly site dependent and that the sp-hybridized carbon atoms are the preferential sites to chemical attacks. Our results also showed that the effectiveness of the oxidation (estimated from the number of oxygen atoms covalently bonded to carbon atoms) follows the α, β, γ-graphyne structure ordering. These differences can be explained by the fact that for α-graphyne structures the oxidation reactions occur in two steps: first, the oxygen atoms are trapped at the center of the large polygonal rings and then they react with the carbon atoms composing of the triple bonds. The small rings of γ-graphyne structures prevent these reactions to occur. The effectiveness of β-graphyne oxidation is between the α- and γ-graphynes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics and structural changes of the oxidation of single-layer graphyne membranes at room temperature. We have considered α, β, and γ-graphyne structures. Our results showed that the oxidation reactions are strongly site dependent and that the sp-hybridized carbon atoms are the preferential sites to chemical attacks. Our results also showed that the effectiveness of the oxidation (estimated from the number of oxygen atoms covalently bonded to carbon atoms) follows the α, β, γ-graphyne structure ordering. These differences can be explained by the fact that for α-graphyne structures the oxidation reactions occur in two steps: first, the oxygen atoms are trapped at the center of the large polygonal rings and then they react with the carbon atoms composing of the triple bonds. The small rings of γ-graphyne structures prevent these reactions to occur. The effectiveness of β-graphyne oxidation is between the α- and γ-graphynes.
Autreto, PA; de Sousa, JM; Galvao, DS
On the Dynamics of Graphdiyne Hydrogenation Journal Article
In: MRS Proceedings, vol. 1549, pp. 59–64, 2013.
@article{Autreto2013,
title = {On the Dynamics of Graphdiyne Hydrogenation},
author = {Autreto, PA and de Sousa, JM and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8915680&fileId=S1946427413006088},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1549},
pages = {59--64},
publisher = {Cambridge University Press},
abstract = {Graphene is a two-dimensional (2D) hexagonal array of carbon atoms in sp2-hybridized states. Graphene presents unique and exceptional electronic, thermal and mechanical properties. However, in its pristine state graphene is a gapless semiconductor, which poses some limitations to its use in some transistor electronics. Because of this there is a renewed interest in other possible two-dimensional carbon-based structures similar to graphene. Examples of this are graphynes and graphdiynes, which are two-dimensional structures, composed of carbon atoms in sp2 and sp-hybridized states. Graphdiynes (benzenoid rings connecting two acetylenic groups) were recently synthesized and they can be intrinsically nonzero gap systems. These systems can be easily hydrogenated and the amount of hydrogenation can be used to tune the band gap value. In this work we have investigated, through fully atomistic molecular dynamics simulations with reactive force field (ReaxFF), the structural and dynamics aspects of the hydrogenation mechanisms of graphdiyne membranes. Our results showed that depending on whether the atoms are in the benzenoid rings or as part of the acetylenic groups, the rates of hydrogenation are quite distinct and change in time in a very complex pattern. Initially, the most probable sites to be hydrogenated are the carbon atoms forming the triple bonds, as expected. But as the amount of hydrogenation increases in time this changes and then the carbon atoms forming single bonds become the preferential sites. The formation of correlated domains observed in hydrogenated graphene is no longer observed in the case of graphdiynes. We have also carried out ab initio DFT calculations for model structures in order to test the reliability of ReaxFF calculations.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Perim, Eric; Paupitz, Ricardo; Autreto, PAS; Galvao, DS
The Hydrogenation Dynamics of h-BN Sheets Journal Article
In: MRS Proceedings, vol. 1549, pp. 91–98, 2013.
@article{perim2013hydrogenation,
title = {The Hydrogenation Dynamics of h-BN Sheets},
author = {Perim, Eric and Paupitz, Ricardo and Autreto, PAS and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8943477&fileId=S1946427413007938},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1549},
pages = {91--98},
publisher = {Cambridge University Press},
abstract = {Hexagonal boron nitride (h-BN), also known as white graphite, is the inorganic analogue of graphite. Single layers of both structures have been already experimentally realized.
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics of hydrogenation of h-BN single-layers membranes.
Our results show that the rate of hydrogenation atoms bonded to the membrane is highly dependent on the temperature and that only at low temperatures there is a preferential bond to boron atoms. Unlike graphanes (hydrogenated graphene), hydrogenated h-BN membranes do not exhibit the formation of correlated domains. Also, the out-of-plane deformations are more pronounced in comparison with the graphene case. After a critical number of incorporated hydrogen atoms the membrane become increasingly defective, lost its two-dimensional character and collapses. The hydrogen radial pair distribution and second-nearest neighbor correlations were also analyzed.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics of hydrogenation of h-BN single-layers membranes.
Our results show that the rate of hydrogenation atoms bonded to the membrane is highly dependent on the temperature and that only at low temperatures there is a preferential bond to boron atoms. Unlike graphanes (hydrogenated graphene), hydrogenated h-BN membranes do not exhibit the formation of correlated domains. Also, the out-of-plane deformations are more pronounced in comparison with the graphene case. After a critical number of incorporated hydrogen atoms the membrane become increasingly defective, lost its two-dimensional character and collapses. The hydrogen radial pair distribution and second-nearest neighbor correlations were also analyzed.
2015
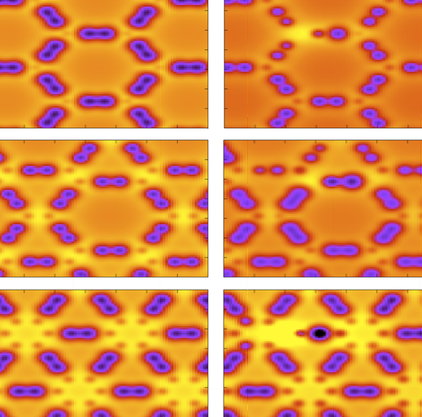
Autreto, Pedro A. S.; Galvao, Douglas S.
Site Dependent Hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation Journal Article
In: Mater. Res. Soc. Symp. Proc. , vol. 1726 , 2015.
Abstract | Links | BibTeX | Tags: graphyne, molecular dynamics, reaxFF
@article{Autreto2015,
title = {Site Dependent Hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Pedro A. S. Autreto and Douglas S. Galvao},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=9702693&fulltextType=RA&fileId=S1946427415004649},
doi = {10.1557/opl.2015.464},
year = {2015},
date = {2015-05-22},
journal = {Mater. Res. Soc. Symp. Proc. },
volume = {1726 },
abstract = {Graphyne is a generic name for a carbon allotrope family of 2D structures, where acetylenic groups connect benzenoid rings, with the coexistence of sp and sp2 hybridized carbon atoms. In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics and structural changes of the hydrogenation of α, β, and γ graphyne forms. Our results showed that the existence of different sites for hydrogen bonding, related to single and triple bonds, makes the process of incorporating hydrogen atoms into graphyne membranes much more complex than the graphene ones. Our results also show that hydrogenation reactions are strongly site dependent and that the sp-hybridized carbon atoms are the preferential sites to chemical attacks. In our cases, the effectiveness of the hydrogenation (estimated from the number of hydrogen atoms covalently bonded to carbon atoms) follows the α, β, γ-graphyne structure ordering.},
keywords = {graphyne, molecular dynamics, reaxFF},
pubstate = {published},
tppubtype = {article}
}
2014
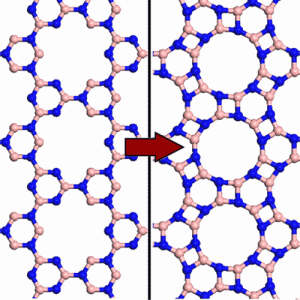
Perim Martins, Eric; Paupitz, Ricardo; Autreto, Pedro Alves da Silva; Galvao, Douglas Soares
Inorganic Graphenylene: A Porous Two-Dimensional Material with Tunable Band Gap Journal Article
In: The Journal of Physical Chemistry C, vol. 118, no. 41, pp. 23670–23674, 2014.
Abstract | Links | BibTeX | Tags: boron nitride, molecular dynamics, porous graphene
@article{perim2014inorganic,
title = {Inorganic Graphenylene: A Porous Two-Dimensional Material with Tunable Band Gap},
author = {Perim Martins, Eric and Paupitz, Ricardo and Autreto, Pedro Alves da Silva and Galvao, Douglas Soares},
url = {http://pubs.acs.org/doi/abs/10.1021/jp502119y},
year = {2014},
date = {2014-01-01},
journal = {The Journal of Physical Chemistry C},
volume = {118},
number = {41},
pages = {23670–23674},
publisher = {American Chemical Society},
abstract = {By means of ab initio calculations, we investigate the possibility of existence of a boron nitride (BN) porous two-dimensional nanosheet, which is geometrically similar to the carbon allotrope known as biphenylene carbon. The proposed structure, which we called inorganic graphenylene (IGP), is formed spontaneously after selective dehydrogenation of the porous boron nitride (BN) structure proposed by Ding et al. We study the structural and electronic properties of both porous BN and IGP, and it is shown that, by selective substitution of B and N atoms with carbon atoms in these structures, the band gap can be significantly reduced, changing their behavior from insulators to semiconductors, thus opening the possibility of band gap engineering for this class of two-dimensional materials.},
keywords = {boron nitride, molecular dynamics, porous graphene},
pubstate = {published},
tppubtype = {article}
}
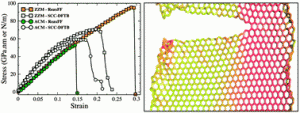
E. Perim T. Botari, P. A. S. Autreto
Mechanical properties and fracture dynamics of silicene membranes Journal Article
In: Phys. Chem. Chem. Phys., vol. 16, pp. 19417–19423, 2014.
Abstract | Links | BibTeX | Tags: mechanical properties, molecular dynamics, silicene
@article{t2014mechanical,
title = {Mechanical properties and fracture dynamics of silicene membranes},
author = {T. Botari, E. Perim, P. A. S. Autreto, A. C. T. van Duin, R. Paupitz, D. S. Galvao},
url = {http://pubs.rsc.org/en/Content/ArticleLanding/2014/CP/C4CP02902J#!divAbstract},
year = {2014},
date = {2014-01-01},
journal = {Phys. Chem. Chem. Phys.},
volume = {16},
pages = {19417--19423},
abstract = {As graphene has become one of the most important materials, there is renewed interest in other similar structures. One example is silicene, the silicon analogue of graphene. It shares some of the remarkable graphene properties, such as the Dirac cone, but presents some distinct ones, such as a pronounced structural buckling. We have investigated, through density functional based tight-binding (DFTB), as well as reactive molecular dynamics (using ReaxFF), the mechanical properties of suspended single-layer silicene. We calculated the elastic constants, analyzed the fracture patterns and edge reconstructions. We also addressed the stress distributions, unbuckling mechanisms and the fracture dependence on the temperature. We analysed the differences due to distinct edge morphologies, namely zigzag and armchair.},
keywords = {mechanical properties, molecular dynamics, silicene},
pubstate = {published},
tppubtype = {article}
}
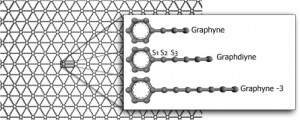
Autreto, PAS; de Sousa, JM; Galvao, DS
Site-dependent hydrogenation on graphdiyne Journal Article
In: Carbon, vol. 77, pp. 829–834, 2014.
Abstract | Links | BibTeX | Tags: graphdiynes, graphynes, molecular dynamics, reaxFF
@article{Autreto2014,
title = {Site-dependent hydrogenation on graphdiyne},
author = {Autreto, PAS and de Sousa, JM and Galvao, DS},
url = {http://www.sciencedirect.com/science/article/pii/S0008622314005429},
year = {2014},
date = {2014-01-01},
journal = {Carbon},
volume = {77},
pages = {829--834},
publisher = {Pergamon},
abstract = {Graphene is one of the most important materials in science today due to its unique and remarkable electronic, thermal and mechanical properties. However in its pristine state, graphene is a gapless semiconductor, what limits its use in transistor electronics. In part due to the revolution created by graphene in materials science, there is a renewed interest in other possible graphene-like two-dimensional structures. Examples of these structures are graphynes and graphdiynes, which are two-dimensional structures, composed of carbon atoms in sp2 and sp-hybridized states. Graphdiynes (benzenoid rings connecting two acetylenic groups) were recently synthesized and some of them are intrinsically nonzero gap systems. These systems can be easily hydrogenated and the relative level of hydrogenation can be used to tune the band gap values. We have investigated, using fully reactive molecular dynamics (ReaxFF), the structural and dynamics aspects of the hydrogenation mechanisms of graphdiyne membranes. Our results showed that the hydrogen bindings have different atom incorporation rates and that the hydrogenation patterns change in time in a very complex way. The formation of correlated domains reported to hydrogenated graphene is no longer observed in graphdiyne cases.},
keywords = {graphdiynes, graphynes, molecular dynamics, reaxFF},
pubstate = {published},
tppubtype = {article}
}

Vinod, Soumya; Tiwary, Chandra Sekhar; da Silva Autreto, Pedro Alves; Taha-Tijerina, Jaime; Ozden, Sehmus; Chipara, Alin Cristian; Vajtai, Robert; Galvao, Douglas S; Narayanan, Tharangattu N; Ajayan, Pulickel M
Low-density three-dimensional foam using self-reinforced hybrid two-dimensional atomic layers Journal Article
In: Nature communications, vol. 5, 2014.
Abstract | Links | BibTeX | Tags: boron nitride, foams, graphene, molecular dynamics
@article{Vinod2014,
title = {Low-density three-dimensional foam using self-reinforced hybrid two-dimensional atomic layers},
author = {Vinod, Soumya and Tiwary, Chandra Sekhar and da Silva Autreto, Pedro Alves and Taha-Tijerina, Jaime and Ozden, Sehmus and Chipara, Alin Cristian and Vajtai, Robert and Galvao, Douglas S and Narayanan, Tharangattu N and Ajayan, Pulickel M},
url = {http://www.nature.com/ncomms/2014/140729/ncomms5541/full/ncomms5541.html},
year = {2014},
date = {2014-01-01},
journal = {Nature communications},
volume = {5},
publisher = {Nature Publishing Group},
abstract = {Low-density nanostructured foams are often limited in applications due to their low mechanical and thermal stabilities. Here we report an approach of building the structural units of three-dimensional (3D) foams using hybrid two-dimensional (2D) atomic layers made of stacked graphene oxide layers reinforced with conformal hexagonal boron nitride (h-BN) platelets. The ultra-low density (1/400 times density of graphite) 3D porous structures are scalably synthesized using solution processing method. A layered 3D foam structure forms due to presence of h-BN and significant improvements in the mechanical properties are observed for the hybrid foam structures, over a range of temperatures, compared with pristine graphene oxide or reduced graphene oxide foams. It is found that domains of h-BN layers on the graphene oxide framework help to reinforce the 2D structural units, providing the observed improvement in mechanical integrity of the 3D foam structure.},
keywords = {boron nitride, foams, graphene, molecular dynamics},
pubstate = {published},
tppubtype = {article}
}
2013

Perim, E; Autreto, PAS; Paupitz, R; Galvao, DS
Dynamical aspects of the unzipping of multiwalled boron nitride nanotubes Journal Article
In: Physical Chemistry Chemical Physics, vol. 15, no. 44, pp. 19147–19150, 2013.
Abstract | BibTeX | Tags: boron nitride, molecular dynamics, reaxFF
@article{perim2013dynamical,
title = {Dynamical aspects of the unzipping of multiwalled boron nitride nanotubes},
author = {Perim, E and Autreto, PAS and Paupitz, R and Galvao, DS},
year = {2013},
date = {2013-01-01},
journal = {Physical Chemistry Chemical Physics},
volume = {15},
number = {44},
pages = {19147--19150},
publisher = {Royal Society of Chemistry},
abstract = {Boron nitride nanoribbons (BNNRs) exhibit very interesting magnetic properties, which could be very useful in the development of spintronic based devices. One possible route to obtain BNNRs is through the unzipping of boron nitride nanotubes (BNNTs), which have been already experimentally realized. In this work, different aspects of the unzipping process of BNNTs were investigated through fully atomistic molecular dynamics simulations using a classical reactive force field (ReaxFF). We investigated multiwalled BNNTs of different diameters and chiralities. Our results show that chirality plays a very important role in the unzipping process, as well as the interlayer coupling. These combined aspects significantly change the fracturing patterns and several other features of the unzipping processes in comparison to the ones observed for carbon nanotubes. Also, similar to carbon nanotubes, defective BNNTs can create regions of very high curvature which can act as a path to the unzipping process.},
keywords = {boron nitride, molecular dynamics, reaxFF},
pubstate = {published},
tppubtype = {article}
}

Paupitz, R; Autreto, Pedro AS; Legoas, SB; Srinivasan, S Goverapet; van Duin, Adri CT; Galvao, DS
Graphene to fluorographene and fluorographane: a theoretical study Journal Article
In: Nanotechnology, vol. 24, no. 3, pp. 035706, 2013.
Abstract | Links | BibTeX | Tags: fluorographane, fluorographene, graphene, molecular dynamics, reaxFF
@article{Paupitz2013,
title = {Graphene to fluorographene and fluorographane: a theoretical study},
author = {Paupitz, R and Autreto, Pedro AS and Legoas, SB and Srinivasan, S Goverapet and van Duin, Adri CT and Galvao, DS},
url = {http://iopscience.iop.org/0957-4484/24/3/035706/article?fromSearchPage=true},
year = {2013},
date = {2013-01-01},
journal = {Nanotechnology},
volume = {24},
number = {3},
pages = {035706},
publisher = {IOP Publishing},
abstract = {We report here a fully reactive molecular dynamics study on the structural and dynamical aspects of the fluorination of graphene membranes (fluorographene). Our results show that fluorination tends to produce defective areas on the graphene membranes with significant distortions of carbon–carbon bonds. Depending on the amount of incorporated fluorine atoms, large membrane holes were observed due to carbon atom losses. These results may explain the broad distribution of the structural lattice parameter values experimentally observed. We have also investigated the effects of mixing hydrogen and fluorine atoms on the graphene functionalization. Our results show that, when in small amounts, the presence of hydrogen atoms produces a significant decrease in the rate of fluorine incorporation onto the membrane. On the other hand, when fluorine is the minority element, it produces a significant catalytic effect on the rate of hydrogen incorporation. We have also observed the spontaneous formation of new hybrid structures with different stable configurations (chair-like, zigzag-like and boat-like) which we named fluorographane.},
keywords = {fluorographane, fluorographene, graphene, molecular dynamics, reaxFF},
pubstate = {published},
tppubtype = {article}
}

Perim, Eric; Santos, Ricardo Paupitz; Autreto, Pedro Alves da Silva; Galvao, Douglas S
Fracture Patterns of Boron Nitride Nanotubes Journal Article
In: MRS Proceedings, vol. 1526, pp. mrsf12–1526, 2013.
BibTeX | Tags: boron nitride, graphene, molecular dynamics
@article{Perim2013,
title = {Fracture Patterns of Boron Nitride Nanotubes},
author = {Perim, Eric and Santos, Ricardo Paupitz and Autreto, Pedro Alves da Silva and Galvao, Douglas S},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1526},
pages = {mrsf12--1526},
publisher = {Cambridge University Press},
keywords = {boron nitride, graphene, molecular dynamics},
pubstate = {published},
tppubtype = {article}
}
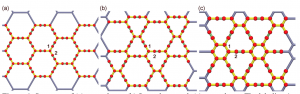
Machado, LD; Autreto, PAS; Galvao, DS
Graphyne Oxidation: Insights From a Reactive Molecular Dynamics Investigation Journal Article
In: MRS Proceedings, vol. 1549, pp. 53–58, 2013.
Abstract | Links | BibTeX | Tags: graphynes, molecular dynamics
@article{Machado2013,
title = {Graphyne Oxidation: Insights From a Reactive Molecular Dynamics Investigation},
author = {Machado, LD and Autreto, PAS and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8963025&fileId=S194642741300941X},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1549},
pages = {53--58},
publisher = {Cambridge University Press},
abstract = {Graphyne is a generic name for a family of carbon allotrope two-dimensional structures where sp2 (single and double bonds) and sp (triple bonds) hybridized states coexists. They exhibit very interesting electronic and mechanical properties sharing some of the unique graphene characteristics. Similarly to graphene, the graphyne electronic properties can be modified by chemical functionalization, such as; hydrogenation, fluorination and oxidation. Oxidation is of particular interest since it can produce significant structural damages.
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics and structural changes of the oxidation of single-layer graphyne membranes at room temperature. We have considered α, β, and γ-graphyne structures. Our results showed that the oxidation reactions are strongly site dependent and that the sp-hybridized carbon atoms are the preferential sites to chemical attacks. Our results also showed that the effectiveness of the oxidation (estimated from the number of oxygen atoms covalently bonded to carbon atoms) follows the α, β, γ-graphyne structure ordering. These differences can be explained by the fact that for α-graphyne structures the oxidation reactions occur in two steps: first, the oxygen atoms are trapped at the center of the large polygonal rings and then they react with the carbon atoms composing of the triple bonds. The small rings of γ-graphyne structures prevent these reactions to occur. The effectiveness of β-graphyne oxidation is between the α- and γ-graphynes.},
keywords = {graphynes, molecular dynamics},
pubstate = {published},
tppubtype = {article}
}
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics and structural changes of the oxidation of single-layer graphyne membranes at room temperature. We have considered α, β, and γ-graphyne structures. Our results showed that the oxidation reactions are strongly site dependent and that the sp-hybridized carbon atoms are the preferential sites to chemical attacks. Our results also showed that the effectiveness of the oxidation (estimated from the number of oxygen atoms covalently bonded to carbon atoms) follows the α, β, γ-graphyne structure ordering. These differences can be explained by the fact that for α-graphyne structures the oxidation reactions occur in two steps: first, the oxygen atoms are trapped at the center of the large polygonal rings and then they react with the carbon atoms composing of the triple bonds. The small rings of γ-graphyne structures prevent these reactions to occur. The effectiveness of β-graphyne oxidation is between the α- and γ-graphynes.
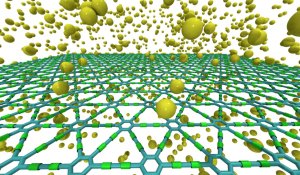
Autreto, PA; de Sousa, JM; Galvao, DS
On the Dynamics of Graphdiyne Hydrogenation Journal Article
In: MRS Proceedings, vol. 1549, pp. 59–64, 2013.
Abstract | Links | BibTeX | Tags: graphynes, molecular dynamics, reaxFF
@article{Autreto2013,
title = {On the Dynamics of Graphdiyne Hydrogenation},
author = {Autreto, PA and de Sousa, JM and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8915680&fileId=S1946427413006088},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1549},
pages = {59--64},
publisher = {Cambridge University Press},
abstract = {Graphene is a two-dimensional (2D) hexagonal array of carbon atoms in sp2-hybridized states. Graphene presents unique and exceptional electronic, thermal and mechanical properties. However, in its pristine state graphene is a gapless semiconductor, which poses some limitations to its use in some transistor electronics. Because of this there is a renewed interest in other possible two-dimensional carbon-based structures similar to graphene. Examples of this are graphynes and graphdiynes, which are two-dimensional structures, composed of carbon atoms in sp2 and sp-hybridized states. Graphdiynes (benzenoid rings connecting two acetylenic groups) were recently synthesized and they can be intrinsically nonzero gap systems. These systems can be easily hydrogenated and the amount of hydrogenation can be used to tune the band gap value. In this work we have investigated, through fully atomistic molecular dynamics simulations with reactive force field (ReaxFF), the structural and dynamics aspects of the hydrogenation mechanisms of graphdiyne membranes. Our results showed that depending on whether the atoms are in the benzenoid rings or as part of the acetylenic groups, the rates of hydrogenation are quite distinct and change in time in a very complex pattern. Initially, the most probable sites to be hydrogenated are the carbon atoms forming the triple bonds, as expected. But as the amount of hydrogenation increases in time this changes and then the carbon atoms forming single bonds become the preferential sites. The formation of correlated domains observed in hydrogenated graphene is no longer observed in the case of graphdiynes. We have also carried out ab initio DFT calculations for model structures in order to test the reliability of ReaxFF calculations.},
keywords = {graphynes, molecular dynamics, reaxFF},
pubstate = {published},
tppubtype = {article}
}
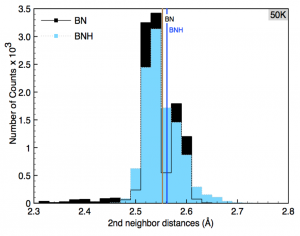
Perim, Eric; Paupitz, Ricardo; Autreto, PAS; Galvao, DS
The Hydrogenation Dynamics of h-BN Sheets Journal Article
In: MRS Proceedings, vol. 1549, pp. 91–98, 2013.
Abstract | Links | BibTeX | Tags: boron nitride, molecular dynamics, reaxFF
@article{perim2013hydrogenation,
title = {The Hydrogenation Dynamics of h-BN Sheets},
author = {Perim, Eric and Paupitz, Ricardo and Autreto, PAS and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8943477&fileId=S1946427413007938},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1549},
pages = {91--98},
publisher = {Cambridge University Press},
abstract = {Hexagonal boron nitride (h-BN), also known as white graphite, is the inorganic analogue of graphite. Single layers of both structures have been already experimentally realized.
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics of hydrogenation of h-BN single-layers membranes.
Our results show that the rate of hydrogenation atoms bonded to the membrane is highly dependent on the temperature and that only at low temperatures there is a preferential bond to boron atoms. Unlike graphanes (hydrogenated graphene), hydrogenated h-BN membranes do not exhibit the formation of correlated domains. Also, the out-of-plane deformations are more pronounced in comparison with the graphene case. After a critical number of incorporated hydrogen atoms the membrane become increasingly defective, lost its two-dimensional character and collapses. The hydrogen radial pair distribution and second-nearest neighbor correlations were also analyzed.},
keywords = {boron nitride, molecular dynamics, reaxFF},
pubstate = {published},
tppubtype = {article}
}
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics of hydrogenation of h-BN single-layers membranes.
Our results show that the rate of hydrogenation atoms bonded to the membrane is highly dependent on the temperature and that only at low temperatures there is a preferential bond to boron atoms. Unlike graphanes (hydrogenated graphene), hydrogenated h-BN membranes do not exhibit the formation of correlated domains. Also, the out-of-plane deformations are more pronounced in comparison with the graphene case. After a critical number of incorporated hydrogen atoms the membrane become increasingly defective, lost its two-dimensional character and collapses. The hydrogen radial pair distribution and second-nearest neighbor correlations were also analyzed.

