
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ
Jaques, Ygor M.; Galvao, Douglas S.
Structural Properties of Nanodroplets Impacting Graphene at High Velocities (accepted) Journal Article
Em: Journal of Molecular Liquids, 2019.
@article{Jaques2019b,
title = {Structural Properties of Nanodroplets Impacting Graphene at High Velocities (accepted)},
author = {Ygor M. Jaques and Douglas S. Galvao},
year = {2019},
date = {2019-02-05},
journal = {Journal of Molecular Liquids},
abstract = {The determination of the wettability of 2D materials is an area of intensive research, as it is decisive on the applications of these systems in nanofluidics. One important part of the wetting characterization is how the spreading of droplets impacting on the surfaces occurs. However, few works address this problem for layered materials. Here, we report a fully atomistic molecular dynamics study on the dynamics of impact of water nanodroplets (100 ̊A of diameter) at high velocities (from 1 up to 15 ̊A/ps) against graphene targets. Our results show that tuning graphene wettability (through parameter changes) significantly affects the structural and dynamical aspects of the nanodroplets. We identified three ranges of velocities with distinct characteristics, from simple deposition of the droplet to spreading with rebound, and finally droplet frag- mentation. We also identify that in an intermediary velocity of 7 ̊A/ps, the pattern of spreading critically changes, due to formation of voids on droplet structure. These voids affect in a detrimental way the droplet spreading on the less hydrophilic surface, as it takes more time to the droplet recover from the spreading and to return to a semi-spherical configuration. When the velocity is increased to values larger than 11 ̊A/ps, the droplet fragments, which reveals the maximum possible spreading.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Ygor M.; Galvao Jaques, Douglas S.
Structural Properties of Nanodroplets Impacting Graphene at High Velocities Online
2018, (Preprint ArXiv:1804.07784).
@online{Jaques2018d,
title = {Structural Properties of Nanodroplets Impacting Graphene at High Velocities},
author = {Jaques, Ygor M.; Galvao, Douglas S.},
url = {https://arxiv.org/abs/1804.07784},
year = {2018},
date = {2018-04-24},
abstract = {We report here a fully atomistic molecular dynamics study on the dynamics of impact of water
nanodroplets (50, 100 and 120 Å of diameter) at high velocity (from 100 up to 1000 m/s) against
graphene targets. Our results show that tuning graphene wettability (through parameter changes)
significantly affects the structural and dynamical aspects of the nanodroplets. We identified three
ranges of velocities with distinct characteristics, from simple deposition of the droplet to
spreading with rebound and finally fragmentation. At Weber numbers lower than 10, the droplets
maintain a steady spreading factor independent of size. After this threshold value, the spread
rapidly grows with increasing Weber numbers. A more hydrophilic graphene surface increases
the spreading values, due to stronger solid-liquid interactions. Nevertheless, droplet size also
influences the fragmentation threshold, as an increased number of molecules make it easier for
the whole droplet overcomes the surface repulsion. },
note = {Preprint ArXiv:1804.07784},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
nanodroplets (50, 100 and 120 Å of diameter) at high velocity (from 100 up to 1000 m/s) against
graphene targets. Our results show that tuning graphene wettability (through parameter changes)
significantly affects the structural and dynamical aspects of the nanodroplets. We identified three
ranges of velocities with distinct characteristics, from simple deposition of the droplet to
spreading with rebound and finally fragmentation. At Weber numbers lower than 10, the droplets
maintain a steady spreading factor independent of size. After this threshold value, the spread
rapidly grows with increasing Weber numbers. A more hydrophilic graphene surface increases
the spreading values, due to stronger solid-liquid interactions. Nevertheless, droplet size also
influences the fragmentation threshold, as an increased number of molecules make it easier for
the whole droplet overcomes the surface repulsion.
Borges, Daiane D; Woellner, Cristiano F; Autreto, Pedro AS; Galvao, Douglas S
2017, (preprint arXiv:1702.00250).
@online{Borges2017,
title = {Water Permeation through Layered Graphene-based Membranes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Borges, Daiane D and Woellner, Cristiano F and Autreto, Pedro AS and Galvao, Douglas S},
url = {https://arxiv.org/abs/1702.00250},
year = {2017},
date = {2017-02-01},
abstract = {Graphene-based membranes have been investigated as promising candidates for water
filtration and gas separation applications. Experimental evidences have shown that graphene
oxide can be impermeable to liquids, vapors and gases, while allowing a fast permeation of water
molecules. This phenomenon has been attributed to the formation of a network of nano
capillaries that allow nearly frictionless water flow while blocking other molecules by steric
hindrance effects. It is supposed that water molecules are transported through the percolated twodimensional
channels formed between graphene-based sheets. Although these channels allow
fast water permeation in such materials, the flow rates are strongly dependent on how the
membranes are fabricated. Also, some fundamental issues regarding the nanoscale mechanisms
of water permeation are still not fully understood and their interpretation remains controversial.
In this work, we have investigated the dynamics of water permeation through pristine graphene
and graphene oxide model membranes. We have carried out fully atomistic classical molecular
dynamics simulations of systems composed of multiple layered graphene-based sheets into
contact with a water reservoir under controlled thermodynamics conditions (e. g., by varying
temperature and pressure values). We have systematically analyzed how the transport dynamics
of the confined nanofluids depend on the interlayer distances and the role of the oxide functional
groups. Our results show the water flux is much more effective for graphene than for graphene
oxide membranes. These results are attributed to the H-bonds formation between oxide
functional groups and water, which traps the water molecules and precludes ultrafast water
transport through the nanochannels.},
note = {preprint arXiv:1702.00250},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
filtration and gas separation applications. Experimental evidences have shown that graphene
oxide can be impermeable to liquids, vapors and gases, while allowing a fast permeation of water
molecules. This phenomenon has been attributed to the formation of a network of nano
capillaries that allow nearly frictionless water flow while blocking other molecules by steric
hindrance effects. It is supposed that water molecules are transported through the percolated twodimensional
channels formed between graphene-based sheets. Although these channels allow
fast water permeation in such materials, the flow rates are strongly dependent on how the
membranes are fabricated. Also, some fundamental issues regarding the nanoscale mechanisms
of water permeation are still not fully understood and their interpretation remains controversial.
In this work, we have investigated the dynamics of water permeation through pristine graphene
and graphene oxide model membranes. We have carried out fully atomistic classical molecular
dynamics simulations of systems composed of multiple layered graphene-based sheets into
contact with a water reservoir under controlled thermodynamics conditions (e. g., by varying
temperature and pressure values). We have systematically analyzed how the transport dynamics
of the confined nanofluids depend on the interlayer distances and the role of the oxide functional
groups. Our results show the water flux is much more effective for graphene than for graphene
oxide membranes. These results are attributed to the H-bonds formation between oxide
functional groups and water, which traps the water molecules and precludes ultrafast water
transport through the nanochannels.
Jaques, Ygor M; Galvao, Douglas S
Nanodroplets Behavior on Graphdiyne Membranes Journal Article
Em: MRS Advances, vol. 2017, pp. 1-6, 2017.
@article{Jaques2017,
title = {Nanodroplets Behavior on Graphdiyne Membranes},
author = {Jaques, Ygor M and Galvao, Douglas S},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/nanodroplets-behavior-on-graphdiyne-membranes/16AD56CAD07570E7F4F194A56E9680C3},
doi = {10.1557/adv.2017.128},
year = {2017},
date = {2017-01-30},
journal = {MRS Advances},
volume = {2017},
pages = {1-6},
abstract = {In this work we have investigated, by fully atomistic reactive (force field ReaxFF) molecular dynamics simulations, some aspects of impact dynamics of water nanodroplets on graphdiyne-like membranes. We simulated graphdiyne-supported membranes impacted by nanodroplets at different velocities (from 100 up to 1500 m/s). The results show that due to the graphdiyne porous and elastic structure, the droplets present an impact dynamics very complex in relation to the ones observed for graphene membranes. Under impact the droplets spread over the surface with a maximum contact radius proportional to the impact velocity. Depending on the energy impact value, a number of water molecules were able to percolate the nanopore sheets. However, even in these cases the droplet shape is preserved and the main differences between the different impact velocities cases reside on the splashing pattern at the maximum spreading.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2019
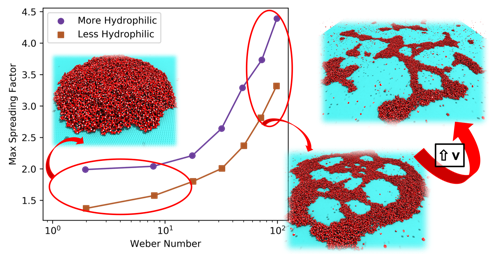
Jaques, Ygor M.; Galvao, Douglas S.
Structural Properties of Nanodroplets Impacting Graphene at High Velocities (accepted) Journal Article
Em: Journal of Molecular Liquids, 2019.
Resumo | BibTeX | Tags: droplets, Graphene, Impact Molecular Dynamics, water
@article{Jaques2019b,
title = {Structural Properties of Nanodroplets Impacting Graphene at High Velocities (accepted)},
author = {Ygor M. Jaques and Douglas S. Galvao},
year = {2019},
date = {2019-02-05},
journal = {Journal of Molecular Liquids},
abstract = {The determination of the wettability of 2D materials is an area of intensive research, as it is decisive on the applications of these systems in nanofluidics. One important part of the wetting characterization is how the spreading of droplets impacting on the surfaces occurs. However, few works address this problem for layered materials. Here, we report a fully atomistic molecular dynamics study on the dynamics of impact of water nanodroplets (100 ̊A of diameter) at high velocities (from 1 up to 15 ̊A/ps) against graphene targets. Our results show that tuning graphene wettability (through parameter changes) significantly affects the structural and dynamical aspects of the nanodroplets. We identified three ranges of velocities with distinct characteristics, from simple deposition of the droplet to spreading with rebound, and finally droplet frag- mentation. We also identify that in an intermediary velocity of 7 ̊A/ps, the pattern of spreading critically changes, due to formation of voids on droplet structure. These voids affect in a detrimental way the droplet spreading on the less hydrophilic surface, as it takes more time to the droplet recover from the spreading and to return to a semi-spherical configuration. When the velocity is increased to values larger than 11 ̊A/ps, the droplet fragments, which reveals the maximum possible spreading.},
keywords = {droplets, Graphene, Impact Molecular Dynamics, water},
pubstate = {published},
tppubtype = {article}
}
2018

Ygor M.; Galvao Jaques, Douglas S.
Structural Properties of Nanodroplets Impacting Graphene at High Velocities Online
2018, (Preprint ArXiv:1804.07784).
Resumo | Links | BibTeX | Tags: droplets, Graphene, Impact Molecular Dynamics, water
@online{Jaques2018d,
title = {Structural Properties of Nanodroplets Impacting Graphene at High Velocities},
author = {Jaques, Ygor M.; Galvao, Douglas S.},
url = {https://arxiv.org/abs/1804.07784},
year = {2018},
date = {2018-04-24},
abstract = {We report here a fully atomistic molecular dynamics study on the dynamics of impact of water
nanodroplets (50, 100 and 120 Å of diameter) at high velocity (from 100 up to 1000 m/s) against
graphene targets. Our results show that tuning graphene wettability (through parameter changes)
significantly affects the structural and dynamical aspects of the nanodroplets. We identified three
ranges of velocities with distinct characteristics, from simple deposition of the droplet to
spreading with rebound and finally fragmentation. At Weber numbers lower than 10, the droplets
maintain a steady spreading factor independent of size. After this threshold value, the spread
rapidly grows with increasing Weber numbers. A more hydrophilic graphene surface increases
the spreading values, due to stronger solid-liquid interactions. Nevertheless, droplet size also
influences the fragmentation threshold, as an increased number of molecules make it easier for
the whole droplet overcomes the surface repulsion. },
note = {Preprint ArXiv:1804.07784},
keywords = {droplets, Graphene, Impact Molecular Dynamics, water},
pubstate = {published},
tppubtype = {online}
}
nanodroplets (50, 100 and 120 Å of diameter) at high velocity (from 100 up to 1000 m/s) against
graphene targets. Our results show that tuning graphene wettability (through parameter changes)
significantly affects the structural and dynamical aspects of the nanodroplets. We identified three
ranges of velocities with distinct characteristics, from simple deposition of the droplet to
spreading with rebound and finally fragmentation. At Weber numbers lower than 10, the droplets
maintain a steady spreading factor independent of size. After this threshold value, the spread
rapidly grows with increasing Weber numbers. A more hydrophilic graphene surface increases
the spreading values, due to stronger solid-liquid interactions. Nevertheless, droplet size also
influences the fragmentation threshold, as an increased number of molecules make it easier for
the whole droplet overcomes the surface repulsion.
2017
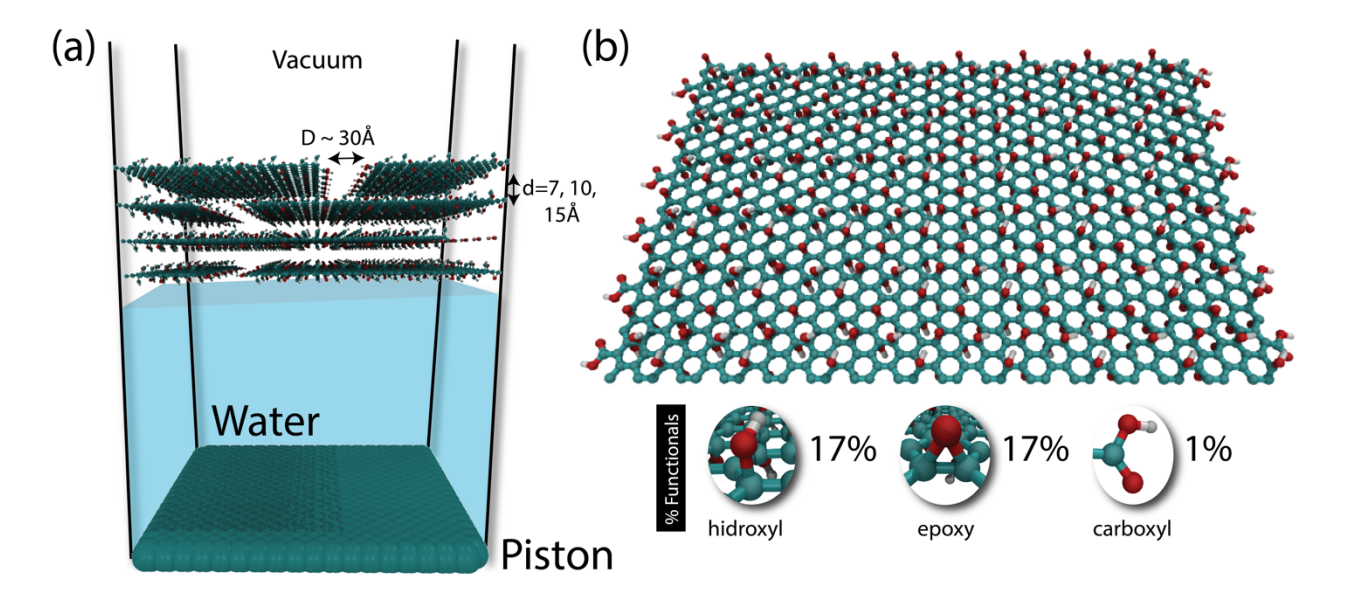
Borges, Daiane D; Woellner, Cristiano F; Autreto, Pedro AS; Galvao, Douglas S
2017, (preprint arXiv:1702.00250).
Resumo | Links | BibTeX | Tags: Graphene, Molecular Dynamics, water
@online{Borges2017,
title = {Water Permeation through Layered Graphene-based Membranes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Borges, Daiane D and Woellner, Cristiano F and Autreto, Pedro AS and Galvao, Douglas S},
url = {https://arxiv.org/abs/1702.00250},
year = {2017},
date = {2017-02-01},
abstract = {Graphene-based membranes have been investigated as promising candidates for water
filtration and gas separation applications. Experimental evidences have shown that graphene
oxide can be impermeable to liquids, vapors and gases, while allowing a fast permeation of water
molecules. This phenomenon has been attributed to the formation of a network of nano
capillaries that allow nearly frictionless water flow while blocking other molecules by steric
hindrance effects. It is supposed that water molecules are transported through the percolated twodimensional
channels formed between graphene-based sheets. Although these channels allow
fast water permeation in such materials, the flow rates are strongly dependent on how the
membranes are fabricated. Also, some fundamental issues regarding the nanoscale mechanisms
of water permeation are still not fully understood and their interpretation remains controversial.
In this work, we have investigated the dynamics of water permeation through pristine graphene
and graphene oxide model membranes. We have carried out fully atomistic classical molecular
dynamics simulations of systems composed of multiple layered graphene-based sheets into
contact with a water reservoir under controlled thermodynamics conditions (e. g., by varying
temperature and pressure values). We have systematically analyzed how the transport dynamics
of the confined nanofluids depend on the interlayer distances and the role of the oxide functional
groups. Our results show the water flux is much more effective for graphene than for graphene
oxide membranes. These results are attributed to the H-bonds formation between oxide
functional groups and water, which traps the water molecules and precludes ultrafast water
transport through the nanochannels.},
note = {preprint arXiv:1702.00250},
keywords = {Graphene, Molecular Dynamics, water},
pubstate = {published},
tppubtype = {online}
}
filtration and gas separation applications. Experimental evidences have shown that graphene
oxide can be impermeable to liquids, vapors and gases, while allowing a fast permeation of water
molecules. This phenomenon has been attributed to the formation of a network of nano
capillaries that allow nearly frictionless water flow while blocking other molecules by steric
hindrance effects. It is supposed that water molecules are transported through the percolated twodimensional
channels formed between graphene-based sheets. Although these channels allow
fast water permeation in such materials, the flow rates are strongly dependent on how the
membranes are fabricated. Also, some fundamental issues regarding the nanoscale mechanisms
of water permeation are still not fully understood and their interpretation remains controversial.
In this work, we have investigated the dynamics of water permeation through pristine graphene
and graphene oxide model membranes. We have carried out fully atomistic classical molecular
dynamics simulations of systems composed of multiple layered graphene-based sheets into
contact with a water reservoir under controlled thermodynamics conditions (e. g., by varying
temperature and pressure values). We have systematically analyzed how the transport dynamics
of the confined nanofluids depend on the interlayer distances and the role of the oxide functional
groups. Our results show the water flux is much more effective for graphene than for graphene
oxide membranes. These results are attributed to the H-bonds formation between oxide
functional groups and water, which traps the water molecules and precludes ultrafast water
transport through the nanochannels.
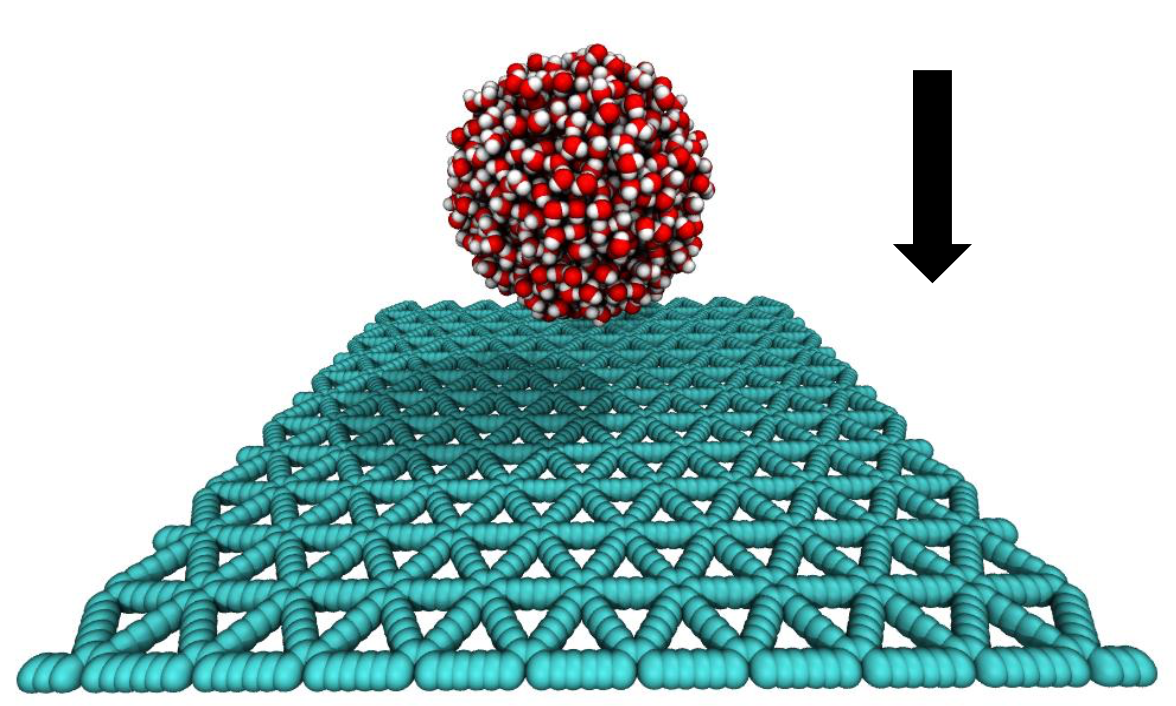
Jaques, Ygor M; Galvao, Douglas S
Nanodroplets Behavior on Graphdiyne Membranes Journal Article
Em: MRS Advances, vol. 2017, pp. 1-6, 2017.
Resumo | Links | BibTeX | Tags: Droplet, graphdiynes, Molecular Dynamics, water
@article{Jaques2017,
title = {Nanodroplets Behavior on Graphdiyne Membranes},
author = {Jaques, Ygor M and Galvao, Douglas S},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/nanodroplets-behavior-on-graphdiyne-membranes/16AD56CAD07570E7F4F194A56E9680C3},
doi = {10.1557/adv.2017.128},
year = {2017},
date = {2017-01-30},
journal = {MRS Advances},
volume = {2017},
pages = {1-6},
abstract = {In this work we have investigated, by fully atomistic reactive (force field ReaxFF) molecular dynamics simulations, some aspects of impact dynamics of water nanodroplets on graphdiyne-like membranes. We simulated graphdiyne-supported membranes impacted by nanodroplets at different velocities (from 100 up to 1500 m/s). The results show that due to the graphdiyne porous and elastic structure, the droplets present an impact dynamics very complex in relation to the ones observed for graphene membranes. Under impact the droplets spread over the surface with a maximum contact radius proportional to the impact velocity. Depending on the energy impact value, a number of water molecules were able to percolate the nanopore sheets. However, even in these cases the droplet shape is preserved and the main differences between the different impact velocities cases reside on the splashing pattern at the maximum spreading.},
keywords = {Droplet, graphdiynes, Molecular Dynamics, water},
pubstate = {published},
tppubtype = {article}
}

