
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ
Eliezer F; Autreto Oliveira, Pedro AS; Woellner
On the mechanical properties of protomene: A theoretical investigation Journal Article
Em: Computational Materials Science, vol. 161, pp. 190-198, 2019.
@article{Oliveira2019c,
title = {On the mechanical properties of protomene: A theoretical investigation},
author = {Oliveira, Eliezer F; Autreto, Pedro AS; Woellner, Cristiano F; Galvao, Douglas S},
year = {2019},
date = {2019-02-07},
journal = {Computational Materials Science},
volume = {161},
pages = {190-198},
abstract = {We report a detailed study through fully atomistic molecular dynamics simulations and DFT calculations on the mechanical properties of protomene. Protomene is a new carbon allotrope composed of a mixture of sp2 and sp3 hybridized states. Our results indicate that protomene presents an anisotropic behavior about tensile deformations. At room temperature, protomene presents an ultimate strength of ~100 GPa and Young's modulus of ~600 GPa, lower than the same for other carbon allotropes. Despite that, protomente presents the highest ultimate strain along the z-direction (~ 24.7%). Our results also show that stretching the protomene along the z-direction or heating it can induce a semiconductor-metallic phase transition, due to a high amount of sp3 bonds that are converted to sp2 ones.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Eliezer F; Autreto Oliveira, Pedro AS; Woellner
Mechanical Properties of Protomene: A Molecular Dynamics Investigation Journal Article
Em: MRS Advances, 2019.
@article{Oliveira2019,
title = {Mechanical Properties of Protomene: A Molecular Dynamics Investigation},
author = {Oliveira, Eliezer F; Autreto, Pedro AS; Woellner, Cristiano F; Galvao, Douglas S},
url = {www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-protomene-a-molecular-dynamics-investigation/CBAC89BDB5942E3353A5C00BD5D0D9CA},
doi = {10.1557/adv.2018.670},
year = {2019},
date = {2019-01-05},
journal = {MRS Advances},
abstract = {Recently, a new class of carbon allotrope called protomene was proposed. This new structure is composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3 carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now, its mechanical properties have not been investigated. In this work, we have investigated protomene mechanical behavior under tensile strain through fully atomistic reactive molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS code. At room temperature, our results show that the protomene is very stable and the obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical fracture.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Pedro AS Autreto Eliezer F Oliveira, Cristiano F Woellner
Mechanical Properties of Protomene: A Molecular Dynamics Investigation Online
2018, (preprint arXiv:1810.09924v1 ).
@online{Oliveira2018g,
title = {Mechanical Properties of Protomene: A Molecular Dynamics Investigation},
author = {Eliezer F Oliveira, Pedro AS Autreto, Cristiano F Woellner, Douglas S Galvao},
url = {https://arxiv.org/abs/1810.09924},
year = {2018},
date = {2018-10-23},
abstract = {Recently, a new class of carbon allotrope called protomene was proposed. This new structure is
composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3
carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations
have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now,
its mechanical properties have not been investigated. In this work, we have investigated
protomene mechanical behavior under tensile strain through fully atomistic reactive
molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS
code. At room temperature, our results show that the protomene is very stable and the
obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest
ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate
strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical
fracture.},
note = {preprint arXiv:1810.09924v1 },
keywords = {},
pubstate = {published},
tppubtype = {online}
}
composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3
carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations
have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now,
its mechanical properties have not been investigated. In this work, we have investigated
protomene mechanical behavior under tensile strain through fully atomistic reactive
molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS
code. At room temperature, our results show that the protomene is very stable and the
obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest
ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate
strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical
fracture.
2019
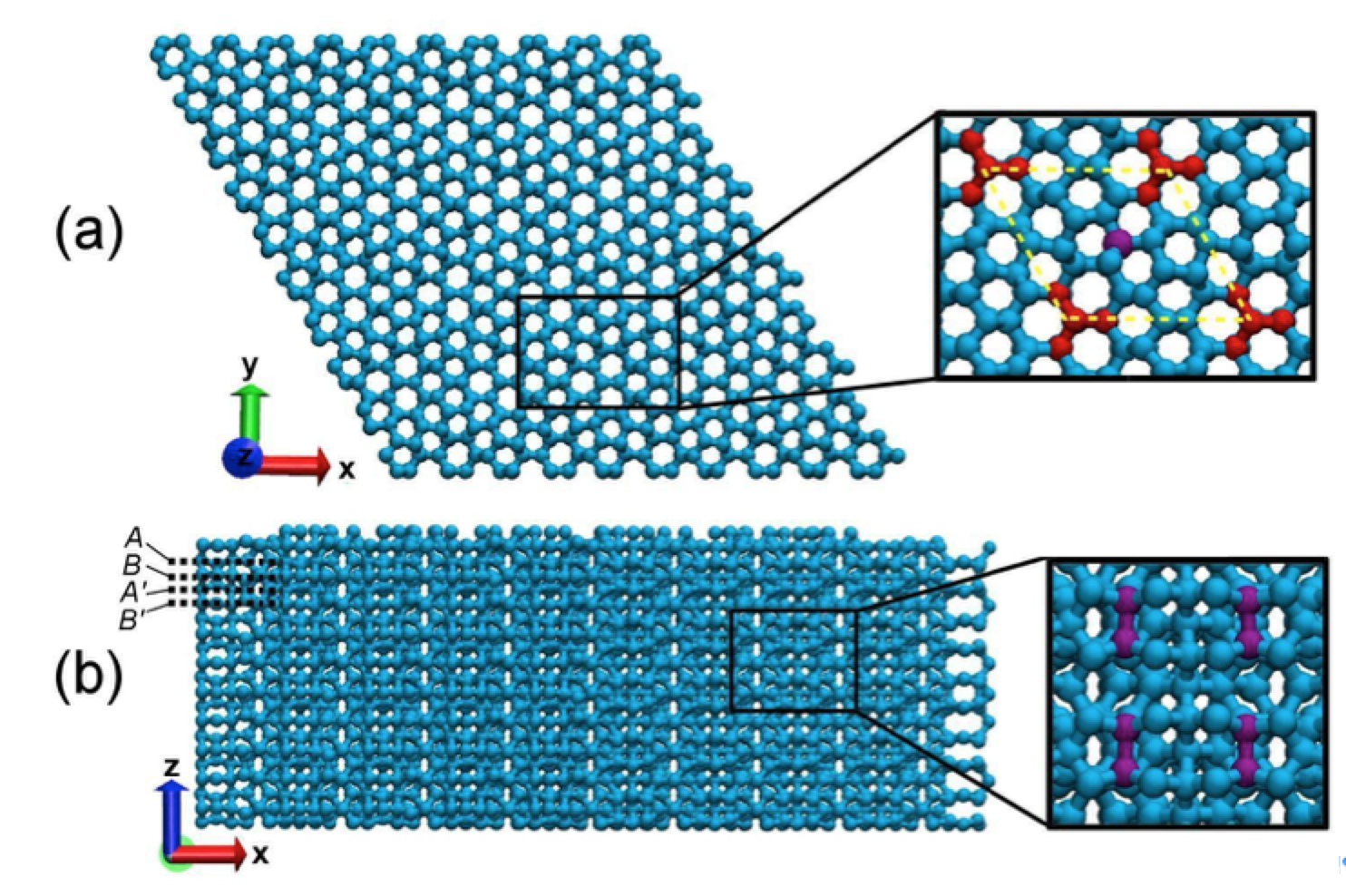
Eliezer F; Autreto Oliveira, Pedro AS; Woellner
On the mechanical properties of protomene: A theoretical investigation Journal Article
Em: Computational Materials Science, vol. 161, pp. 190-198, 2019.
Resumo | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, protomene
@article{Oliveira2019c,
title = {On the mechanical properties of protomene: A theoretical investigation},
author = {Oliveira, Eliezer F; Autreto, Pedro AS; Woellner, Cristiano F; Galvao, Douglas S},
year = {2019},
date = {2019-02-07},
journal = {Computational Materials Science},
volume = {161},
pages = {190-198},
abstract = {We report a detailed study through fully atomistic molecular dynamics simulations and DFT calculations on the mechanical properties of protomene. Protomene is a new carbon allotrope composed of a mixture of sp2 and sp3 hybridized states. Our results indicate that protomene presents an anisotropic behavior about tensile deformations. At room temperature, protomene presents an ultimate strength of ~100 GPa and Young's modulus of ~600 GPa, lower than the same for other carbon allotropes. Despite that, protomente presents the highest ultimate strain along the z-direction (~ 24.7%). Our results also show that stretching the protomene along the z-direction or heating it can induce a semiconductor-metallic phase transition, due to a high amount of sp3 bonds that are converted to sp2 ones.},
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, protomene},
pubstate = {published},
tppubtype = {article}
}
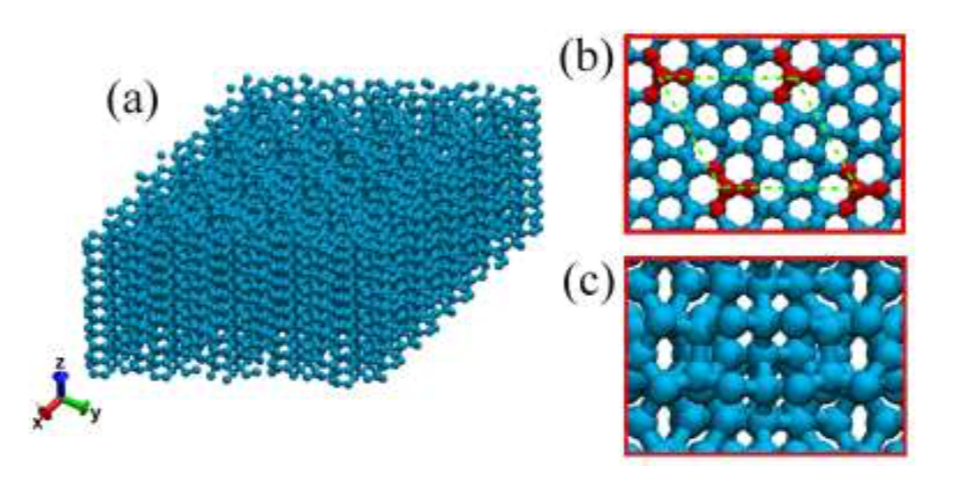
Eliezer F; Autreto Oliveira, Pedro AS; Woellner
Mechanical Properties of Protomene: A Molecular Dynamics Investigation Journal Article
Em: MRS Advances, 2019.
Resumo | Links | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, protomene
@article{Oliveira2019,
title = {Mechanical Properties of Protomene: A Molecular Dynamics Investigation},
author = {Oliveira, Eliezer F; Autreto, Pedro AS; Woellner, Cristiano F; Galvao, Douglas S},
url = {www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-protomene-a-molecular-dynamics-investigation/CBAC89BDB5942E3353A5C00BD5D0D9CA},
doi = {10.1557/adv.2018.670},
year = {2019},
date = {2019-01-05},
journal = {MRS Advances},
abstract = {Recently, a new class of carbon allotrope called protomene was proposed. This new structure is composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3 carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now, its mechanical properties have not been investigated. In this work, we have investigated protomene mechanical behavior under tensile strain through fully atomistic reactive molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS code. At room temperature, our results show that the protomene is very stable and the obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical fracture.
},
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, protomene},
pubstate = {published},
tppubtype = {article}
}
2018
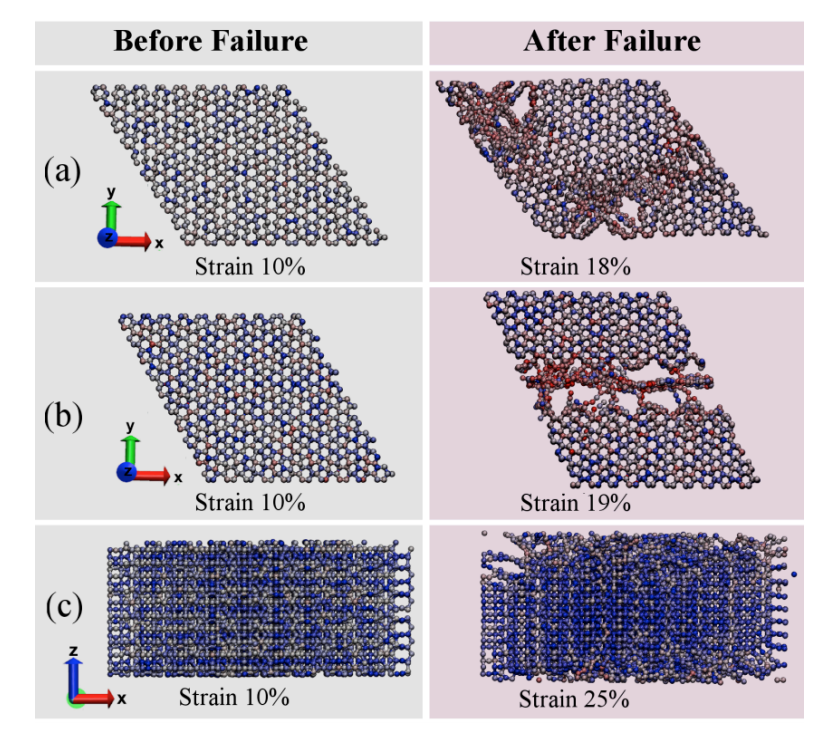
Pedro AS Autreto Eliezer F Oliveira, Cristiano F Woellner
Mechanical Properties of Protomene: A Molecular Dynamics Investigation Online
2018, (preprint arXiv:1810.09924v1 ).
Resumo | Links | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, protomene
@online{Oliveira2018g,
title = {Mechanical Properties of Protomene: A Molecular Dynamics Investigation},
author = {Eliezer F Oliveira, Pedro AS Autreto, Cristiano F Woellner, Douglas S Galvao},
url = {https://arxiv.org/abs/1810.09924},
year = {2018},
date = {2018-10-23},
abstract = {Recently, a new class of carbon allotrope called protomene was proposed. This new structure is
composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3
carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations
have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now,
its mechanical properties have not been investigated. In this work, we have investigated
protomene mechanical behavior under tensile strain through fully atomistic reactive
molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS
code. At room temperature, our results show that the protomene is very stable and the
obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest
ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate
strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical
fracture.},
note = {preprint arXiv:1810.09924v1 },
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, protomene},
pubstate = {published},
tppubtype = {online}
}
composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3
carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations
have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now,
its mechanical properties have not been investigated. In this work, we have investigated
protomene mechanical behavior under tensile strain through fully atomistic reactive
molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS
code. At room temperature, our results show that the protomene is very stable and the
obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest
ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate
strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical
fracture.

