
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ
de Sousa, Jose Moreira; Autreto, Pedro da Silva; Galvao, Douglas Soares
Hydrogenation Dynamics Process of Single-wall Carbon Nanotube Twisted (under review) Journal Article
Em: 2019.
@article{deSousa2019d,
title = {Hydrogenation Dynamics Process of Single-wall Carbon Nanotube Twisted (under review)},
author = {de Sousa, Jose Moreira and Autreto, Pedro da Silva and Galvao, Douglas Soares},
year = {2019},
date = {2019-07-15},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Splugues, Vinicius; da Silva Autreto, Pedro Alves; Galvao, Douglas S
Hydrogenation Dynamics of Biphenylene Carbon (Graphenylene) Membranes Journal Article
Em: MRS Advances, vol. 2017, pp. 1-6, 2017.
@article{Splugues2017,
title = {Hydrogenation Dynamics of Biphenylene Carbon (Graphenylene) Membranes},
author = {Splugues, Vinicius and da Silva Autreto, Pedro Alves and Galvao, Douglas S},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/hydrogenation-dynamics-of-biphenylene-carbon-graphenylene-membranes/139DB900D41560D64F352A31CE219D3A},
doi = {10.1557/adv.2017.239},
year = {2017},
date = {2017-02-28},
journal = {MRS Advances},
volume = {2017},
pages = {1-6},
abstract = {The advent of graphene created a revolution in materials science. Because of this there is a renewed interest in other carbon-based structures. Graphene is the ultimate (just one atom thick) membrane. It has been proposed that graphene can work as impermeable membrane to standard gases, such argon and helium. Graphene-like porous membranes, but presenting larger porosity and potential selectivity would have many technological applications. Biphenylene carbon (BPC), sometimes called graphenylene, is one of these structures. BPC is a porous two-dimensional (planar) allotrope carbon, with its pores resembling typical sieve cavities and/or some kind of zeolites. In this work, we have investigated the hydrogenation dynamics of BPC membranes under different conditions (hydrogenation plasma density, temperature, etc.). We have carried out an extensive study through fully atomistic molecular dynamics (MD) simulations using the reactive force field ReaxFF, as implemented in the well-known Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) code. Our results show that the BPC hydrogenation processes exhibit very complex patterns and the formation of correlated domains (hydrogenated islands) observed in the case of graphene hydrogenation was also observed here. MD results also show that under hydrogenation BPC structure undergoes a change in its topology, the pores undergoing structural transformations and extensive hydrogenation can produce significant structural damages, with the formation of large defective areas and large structural holes, leading to structural collapse.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
de Sousa, Jose M.; Autreto, Pedro A. S.; Galvao, Douglas S.
Hydrogenation Dynamics of Twisted Carbon Nanotubes Online
2015, (ArXiv preprint).
@online{deSousa2015,
title = {Hydrogenation Dynamics of Twisted Carbon Nanotubes},
author = {Jose M. de Sousa and Pedro A. S. Autreto and Douglas S. Galvao},
url = {http://arxiv.org/abs/1510.00265},
year = {2015},
date = {2015-10-01},
abstract = {Carbon Nanotubes (CNTs) are one of the most important materials in nanotechnology. In some of their technological applications (electromechanical oscillators and mechanical actuators for artificial muscles, for instance), it is necessary to subject them to large deformations. Although this frequently happens in air, there are only few studies about the interaction of deformed CNTs with the atmosphere and the dynamics of these processes has not yet been addressed. In this work, we have investigated, through fully atomistic reactive molecular dynamics simulations, the process of hydrogenation of highly twisted CNTs. Our results show that hydrogenation effective ratio is directly related to the tube twist angle values and can lead to twisted tube fractures with well defined patterns (unzip-like). Our results also show that these fracture processes can be exploited to controllably produce graphene nanoribbons.},
note = {ArXiv preprint},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Pedro A. S. Autreto, Douglas S. Galvao
Site dependent hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation Online
2015, (ArXiv draft of MRS Proceedings, 1726, mrsf14-1726-j02-02 (2015)).
@online{Autreto2015,
title = {Site dependent hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Pedro A. S. Autreto, Douglas S. Galvao},
url = {http://arxiv.org/abs/1501.04521},
year = {2015},
date = {2015-01-19},
journal = {arXiv preprint 1501.04521},
abstract = {Graphyne is a generic name for a carbon allotrope family of 2D structures, where acetylenic groups connect benzenoid rings, with the coexistence of sp and sp2 hybridized carbon atoms. In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics and structural changes of the hydrogenation of ALPHA, BETA, GAMMA graphyne forms. Our results showed that the existence of different sites for hydrogen bonding, related to single and triple bonds, makes the process of incorporating hydrogen atoms into graphyne membranes much more complex than the graphene ones. Our results also show that hydrogenation reactions are strongly site dependent and that the sp-hybridized carbon atoms are the preferential sites to chemical attacks. In our cases, the effectiveness of the hydrogenation (estimated from the number of hydrogen atoms covalently bonded to carbon atoms) follows the ALPHA, BETA, GAMMA graphyne structure ordering.},
note = {ArXiv draft of MRS Proceedings, 1726, mrsf14-1726-j02-02 (2015)},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Pedro A. S. Autreto, Douglas S. Galvao
Site Dependent Hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation Proceedings
vol. 1726, não mrsf14-1726-j02-02, 2015, (MRS Proceedings, 1726, mrsf14-1726-j02-02 ).
@proceedings{Autreto2015b,
title = {Site Dependent Hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Pedro A. S. Autreto, Douglas S. Galvao},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=9702693&fulltextType=RA&fileId=S1946427415004649},
doi = {10.1557/opl.2015.464},
year = {2015},
date = {2015-01-01},
journal = {Mater. Res. Soc. Symp. Proc. },
volume = {1726},
number = {mrsf14-1726-j02-02},
abstract = {Graphyne is a generic name for a carbon allotrope family of 2D structures, where acetylenic groups connect benzenoid rings, with the coexistence of sp and sp2 hybridized carbon atoms. In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics and structural changes of the hydrogenation of α, β, and γ graphyne forms. Our results showed that the existence of different sites for hydrogen bonding, related to single and triple bonds, makes the process of incorporating hydrogen atoms into graphyne membranes much more complex than the graphene ones. Our results also show that hydrogenation reactions are strongly site dependent and that the sp-hybridized carbon atoms are the preferential sites to chemical attacks. In our cases, the effectiveness of the hydrogenation (estimated from the number of hydrogen atoms covalently bonded to carbon atoms) follows the α, β, γ-graphyne structure ordering.},
note = {MRS Proceedings, 1726, mrsf14-1726-j02-02 },
keywords = {},
pubstate = {published},
tppubtype = {proceedings}
}
Autreto, PA; de Sousa, JM; Galvao, DS
On the Dynamics of Graphdiyne Hydrogenation Proceedings
Cambridge University Press, vol. 1549, 2013.
@proceedings{autreto2013dynamics,
title = {On the Dynamics of Graphdiyne Hydrogenation},
author = {Autreto, PA and de Sousa, JM and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8915680&fileId=S1946427413006088},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1549},
pages = {59--64},
publisher = {Cambridge University Press},
abstract = {Graphene is a two-dimensional (2D) hexagonal array of carbon atoms in sp2-hybridized states. Graphene presents unique and exceptional electronic, thermal and mechanical properties. However, in its pristine state graphene is a gapless semiconductor, which poses some limitations to its use in some transistor electronics. Because of this there is a renewed interest in other possible two-dimensional carbon-based structures similar to graphene. Examples of this are graphynes and graphdiynes, which are two-dimensional structures, composed of carbon atoms in sp2 and sp-hybridized states. Graphdiynes (benzenoid rings connecting two acetylenic groups) were recently synthesized and they can be intrinsically nonzero gap systems. These systems can be easily hydrogenated and the amount of hydrogenation can be used to tune the band gap value. In this work we have investigated, through fully atomistic molecular dynamics simulations with reactive force field (ReaxFF), the structural and dynamics aspects of the hydrogenation mechanisms of graphdiyne membranes. Our results showed that depending on whether the atoms are in the benzenoid rings or as part of the acetylenic groups, the rates of hydrogenation are quite distinct and change in time in a very complex pattern. Initially, the most probable sites to be hydrogenated are the carbon atoms forming the triple bonds, as expected. But as the amount of hydrogenation increases in time this changes and then the carbon atoms forming single bonds become the preferential sites. The formation of correlated domains observed in hydrogenated graphene is no longer observed in the case of graphdiynes. We have also carried out ab initio DFT calculations for model structures in order to test the reliability of ReaxFF calculations.},
keywords = {},
pubstate = {published},
tppubtype = {proceedings}
}
Perim, Eric; Paupitz, Ricardo; Autreto, PAS; Galvao, DS
The Hydrogenation Dynamics of h-BN Sheets Proceedings
Cambridge University Press, vol. 1549, 2013.
@proceedings{perim2013hydrogenation,
title = {The Hydrogenation Dynamics of h-BN Sheets},
author = {Perim, Eric and Paupitz, Ricardo and Autreto, PAS and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8943477&fileId=S1946427413007938},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1549},
pages = {91--98},
publisher = {Cambridge University Press},
abstract = {Hexagonal boron nitride (h-BN), also known as white graphite, is the inorganic analogue of graphite. Single layers of both structures have been already experimentally realized.
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics of hydrogenation of h-BN single-layers membranes.
Our results show that the rate of hydrogenation atoms bonded to the membrane is highly dependent on the temperature and that only at low temperatures there is a preferential bond to boron atoms. Unlike graphanes (hydrogenated graphene), hydrogenated h-BN membranes do not exhibit the formation of correlated domains. Also, the out-of-plane deformations are more pronounced in comparison with the graphene case. After a critical number of incorporated hydrogen atoms the membrane become increasingly defective, lost its two-dimensional character and collapses. The hydrogen radial pair distribution and second-nearest neighbor correlations were also analyzed.},
keywords = {},
pubstate = {published},
tppubtype = {proceedings}
}
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics of hydrogenation of h-BN single-layers membranes.
Our results show that the rate of hydrogenation atoms bonded to the membrane is highly dependent on the temperature and that only at low temperatures there is a preferential bond to boron atoms. Unlike graphanes (hydrogenated graphene), hydrogenated h-BN membranes do not exhibit the formation of correlated domains. Also, the out-of-plane deformations are more pronounced in comparison with the graphene case. After a critical number of incorporated hydrogen atoms the membrane become increasingly defective, lost its two-dimensional character and collapses. The hydrogen radial pair distribution and second-nearest neighbor correlations were also analyzed.
Autreto, Pedro AS; Flores, Marcelo Z; Legoas, Sergio B; Santos, Ricardo PB; Galvao, Douglas S
Cambridge University Press, vol. 1284, 2011.
@proceedings{autreto2011fully,
title = {A Fully Atomistic Reactive Molecular Dynamics Study on the Formation of Graphane from Graphene Hydrogenated Membranes.},
author = {Autreto, Pedro AS and Flores, Marcelo Z and Legoas, Sergio B and Santos, Ricardo PB and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8364784&fileId=S1946427411013583},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1284},
pages = {mrsf10--1284},
publisher = {Cambridge University Press},
abstract = {Using fully reactive molecular dynamics methodologies we investigated the structural and dynamical aspects of the fluorination mechanism leading to fluorographene formation from graphene membranes. Fluorination tends to produce significant defective areas on the membranes with variation on the typical carbon-carbon distances, sometimes with the presence of large holes due to carbon losses. The results obtained in our simulations are in good agreement with the broad distribution of values for the lattice parameter experimentally observed. We have also investigated mixed atmospheres composed by H and F atoms. When H is present in small quantities an expressive reduction on the rate of incorporation of fluorine was observed. On the other hand when fluorine atoms are present in small quantities in a hydrogen atmosphere, they induce an increasing on the hydrogen incorporation and the formation of locally self-organized structure of adsorbed H and F atoms.},
keywords = {},
pubstate = {published},
tppubtype = {proceedings}
}
Flores, Marcelo ZS; Autreto, Pedro AS; Legoas, Sergio B; Galvao, Douglas S
Graphene to graphane: a theoretical study Journal Article
Em: Nanotechnology, vol. 20, não 46, pp. 465704, 2009.
@article{flores2009graphene,
title = {Graphene to graphane: a theoretical study},
author = {Flores, Marcelo ZS and Autreto, Pedro AS and Legoas, Sergio B and Galvao, Douglas S},
url = {http://iopscience.iop.org/0957-4484/20/46/465704},
year = {2009},
date = {2009-01-01},
journal = {Nanotechnology},
volume = {20},
number = {46},
pages = {465704},
publisher = {IOP Publishing},
abstract = {Graphane is a two-dimensional system consisting of a single layer of fully saturated (sp3 hybridization) carbon atoms. In an ideal graphane structure C–H bonds exhibit an alternating pattern (up and down with relation to the plane defined by the carbon atoms). In this work we have investigated, using ab initio and reactive molecular dynamics simulations, the role of H frustration (breaking the H atoms' up and down alternating pattern) in graphane-like structures. Our results show that a significant percentage of uncorrelated H frustrated domains are formed in the early stages of the hydrogenation process leading to membrane shrinkage and extensive membrane corrugations. These results also suggest that large domains of perfect graphane-like structures are unlikely to be formed, as H frustrated domains are always present.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Legoas, Sergio B; Autreto, Pedro AS; Flores, Marcelo ZS; Galvao, Douglas S
Graphene to graphane: the role of H frustration in lattice contraction Journal Article
Em: arXiv preprint arXiv:0903.0278, 2009.
@article{legoas2009graphene,
title = {Graphene to graphane: the role of H frustration in lattice contraction},
author = {Legoas, Sergio B and Autreto, Pedro AS and Flores, Marcelo ZS and Galvao, Douglas S},
url = {http://arxiv.org/abs/0903.0278},
year = {2009},
date = {2009-01-01},
journal = {arXiv preprint arXiv:0903.0278},
abstract = {Graphane is a two-dimensional system consisting of a single planar layer of fully saturated (sp3 hybridization) carbon atoms with H atoms attached to them in an alternating pattern (up and down with relation to the plane defined by the carbon atoms). Stable graphane structures were theoretically predicted to exist some years ago and just experimentally realized through hydrogenation of graphene membranes. In this work we have investigated using textit{ab initio} and reactive molecular dynamics the role of H frustration (breaking the H atoms up and down alternating pattern) in graphane-like structures. Our results show that H frustration significantly contributes to lattice contraction. The dynamical aspects of converting graphene to graphane is also addressed.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2019
de Sousa, Jose Moreira; Autreto, Pedro da Silva; Galvao, Douglas Soares
Hydrogenation Dynamics Process of Single-wall Carbon Nanotube Twisted (under review) Journal Article
Em: 2019.
BibTeX | Tags: Carbon Nanotubes, Hydrogenation, Mechanical Properties, Molecular Dynamics
@article{deSousa2019d,
title = {Hydrogenation Dynamics Process of Single-wall Carbon Nanotube Twisted (under review)},
author = {de Sousa, Jose Moreira and Autreto, Pedro da Silva and Galvao, Douglas Soares},
year = {2019},
date = {2019-07-15},
keywords = {Carbon Nanotubes, Hydrogenation, Mechanical Properties, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
2017
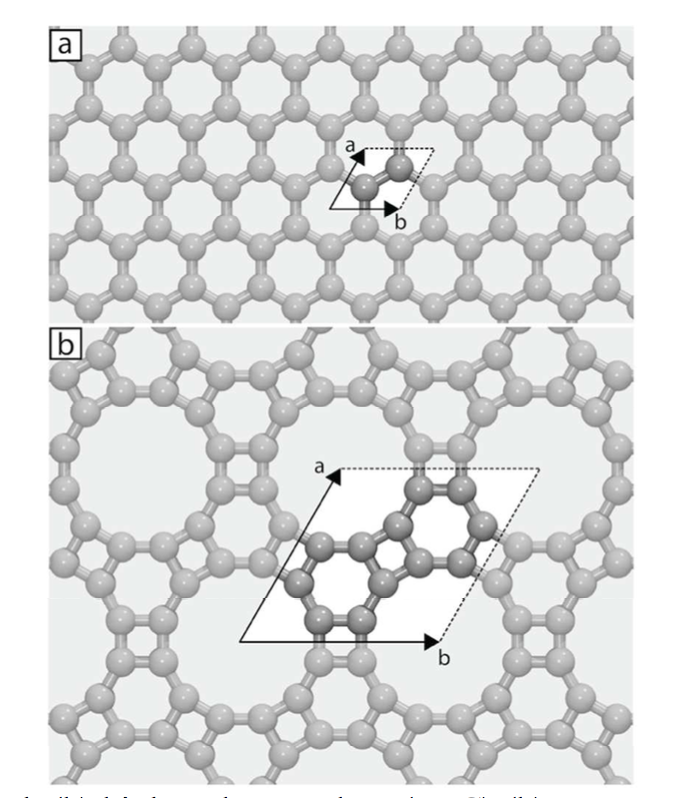
Splugues, Vinicius; da Silva Autreto, Pedro Alves; Galvao, Douglas S
Hydrogenation Dynamics of Biphenylene Carbon (Graphenylene) Membranes Journal Article
Em: MRS Advances, vol. 2017, pp. 1-6, 2017.
Resumo | Links | BibTeX | Tags: Graphene, Hydrogenation, Molecular Dynamics
@article{Splugues2017,
title = {Hydrogenation Dynamics of Biphenylene Carbon (Graphenylene) Membranes},
author = {Splugues, Vinicius and da Silva Autreto, Pedro Alves and Galvao, Douglas S},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/hydrogenation-dynamics-of-biphenylene-carbon-graphenylene-membranes/139DB900D41560D64F352A31CE219D3A},
doi = {10.1557/adv.2017.239},
year = {2017},
date = {2017-02-28},
journal = {MRS Advances},
volume = {2017},
pages = {1-6},
abstract = {The advent of graphene created a revolution in materials science. Because of this there is a renewed interest in other carbon-based structures. Graphene is the ultimate (just one atom thick) membrane. It has been proposed that graphene can work as impermeable membrane to standard gases, such argon and helium. Graphene-like porous membranes, but presenting larger porosity and potential selectivity would have many technological applications. Biphenylene carbon (BPC), sometimes called graphenylene, is one of these structures. BPC is a porous two-dimensional (planar) allotrope carbon, with its pores resembling typical sieve cavities and/or some kind of zeolites. In this work, we have investigated the hydrogenation dynamics of BPC membranes under different conditions (hydrogenation plasma density, temperature, etc.). We have carried out an extensive study through fully atomistic molecular dynamics (MD) simulations using the reactive force field ReaxFF, as implemented in the well-known Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) code. Our results show that the BPC hydrogenation processes exhibit very complex patterns and the formation of correlated domains (hydrogenated islands) observed in the case of graphene hydrogenation was also observed here. MD results also show that under hydrogenation BPC structure undergoes a change in its topology, the pores undergoing structural transformations and extensive hydrogenation can produce significant structural damages, with the formation of large defective areas and large structural holes, leading to structural collapse.
},
keywords = {Graphene, Hydrogenation, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
2015
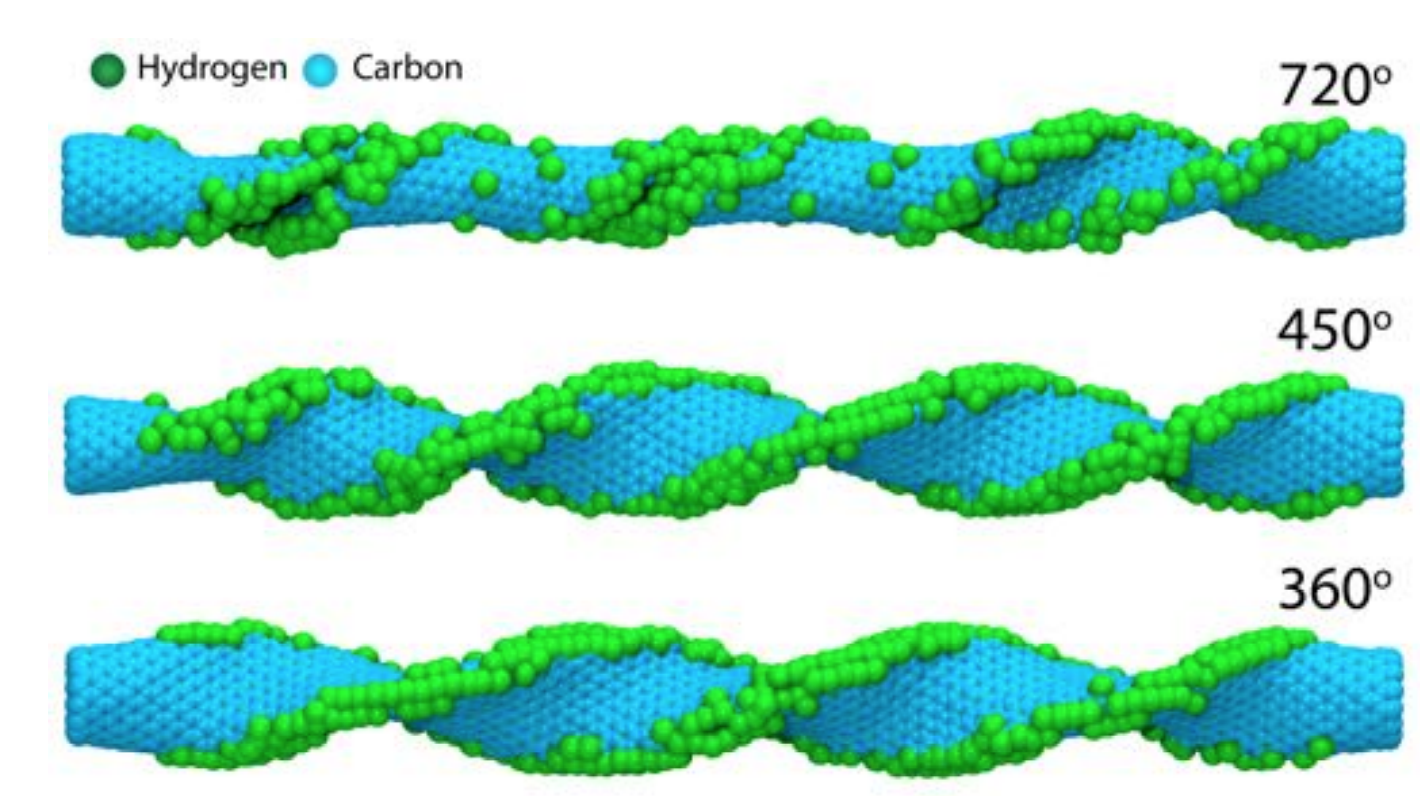
de Sousa, Jose M.; Autreto, Pedro A. S.; Galvao, Douglas S.
Hydrogenation Dynamics of Twisted Carbon Nanotubes Online
2015, (ArXiv preprint).
Resumo | Links | BibTeX | Tags: Carbon Nanotubes, Hydrogenation, Mechanical Properties, Molecular Dynamics
@online{deSousa2015,
title = {Hydrogenation Dynamics of Twisted Carbon Nanotubes},
author = {Jose M. de Sousa and Pedro A. S. Autreto and Douglas S. Galvao},
url = {http://arxiv.org/abs/1510.00265},
year = {2015},
date = {2015-10-01},
abstract = {Carbon Nanotubes (CNTs) are one of the most important materials in nanotechnology. In some of their technological applications (electromechanical oscillators and mechanical actuators for artificial muscles, for instance), it is necessary to subject them to large deformations. Although this frequently happens in air, there are only few studies about the interaction of deformed CNTs with the atmosphere and the dynamics of these processes has not yet been addressed. In this work, we have investigated, through fully atomistic reactive molecular dynamics simulations, the process of hydrogenation of highly twisted CNTs. Our results show that hydrogenation effective ratio is directly related to the tube twist angle values and can lead to twisted tube fractures with well defined patterns (unzip-like). Our results also show that these fracture processes can be exploited to controllably produce graphene nanoribbons.},
note = {ArXiv preprint},
keywords = {Carbon Nanotubes, Hydrogenation, Mechanical Properties, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}
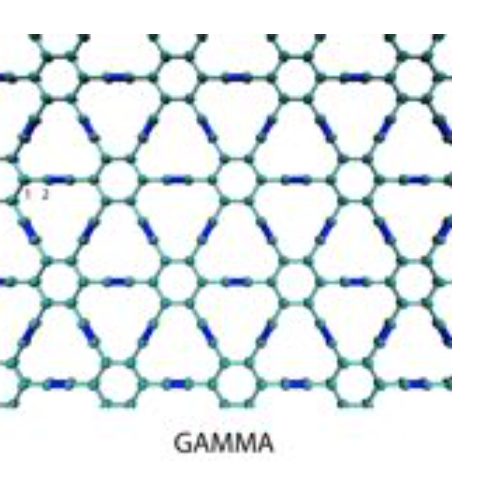
Pedro A. S. Autreto, Douglas S. Galvao
Site dependent hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation Online
2015, (ArXiv draft of MRS Proceedings, 1726, mrsf14-1726-j02-02 (2015)).
Resumo | Links | BibTeX | Tags: Graphynes, Hydrogenation, Molecular Dynamics
@online{Autreto2015,
title = {Site dependent hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Pedro A. S. Autreto, Douglas S. Galvao},
url = {http://arxiv.org/abs/1501.04521},
year = {2015},
date = {2015-01-19},
journal = {arXiv preprint 1501.04521},
abstract = {Graphyne is a generic name for a carbon allotrope family of 2D structures, where acetylenic groups connect benzenoid rings, with the coexistence of sp and sp2 hybridized carbon atoms. In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics and structural changes of the hydrogenation of ALPHA, BETA, GAMMA graphyne forms. Our results showed that the existence of different sites for hydrogen bonding, related to single and triple bonds, makes the process of incorporating hydrogen atoms into graphyne membranes much more complex than the graphene ones. Our results also show that hydrogenation reactions are strongly site dependent and that the sp-hybridized carbon atoms are the preferential sites to chemical attacks. In our cases, the effectiveness of the hydrogenation (estimated from the number of hydrogen atoms covalently bonded to carbon atoms) follows the ALPHA, BETA, GAMMA graphyne structure ordering.},
note = {ArXiv draft of MRS Proceedings, 1726, mrsf14-1726-j02-02 (2015)},
keywords = {Graphynes, Hydrogenation, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}
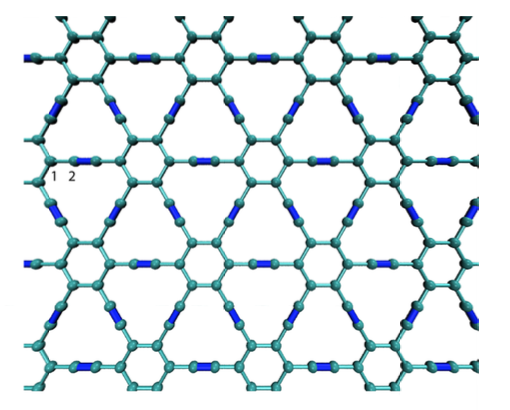
Pedro A. S. Autreto, Douglas S. Galvao
Site Dependent Hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation Proceedings
vol. 1726, não mrsf14-1726-j02-02, 2015, (MRS Proceedings, 1726, mrsf14-1726-j02-02 ).
Resumo | Links | BibTeX | Tags: Graphynes, Hydrogenation, Molecular Dynamics
@proceedings{Autreto2015b,
title = {Site Dependent Hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Pedro A. S. Autreto, Douglas S. Galvao},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=9702693&fulltextType=RA&fileId=S1946427415004649},
doi = {10.1557/opl.2015.464},
year = {2015},
date = {2015-01-01},
journal = {Mater. Res. Soc. Symp. Proc. },
volume = {1726},
number = {mrsf14-1726-j02-02},
abstract = {Graphyne is a generic name for a carbon allotrope family of 2D structures, where acetylenic groups connect benzenoid rings, with the coexistence of sp and sp2 hybridized carbon atoms. In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics and structural changes of the hydrogenation of α, β, and γ graphyne forms. Our results showed that the existence of different sites for hydrogen bonding, related to single and triple bonds, makes the process of incorporating hydrogen atoms into graphyne membranes much more complex than the graphene ones. Our results also show that hydrogenation reactions are strongly site dependent and that the sp-hybridized carbon atoms are the preferential sites to chemical attacks. In our cases, the effectiveness of the hydrogenation (estimated from the number of hydrogen atoms covalently bonded to carbon atoms) follows the α, β, γ-graphyne structure ordering.},
note = {MRS Proceedings, 1726, mrsf14-1726-j02-02 },
keywords = {Graphynes, Hydrogenation, Molecular Dynamics},
pubstate = {published},
tppubtype = {proceedings}
}
2013
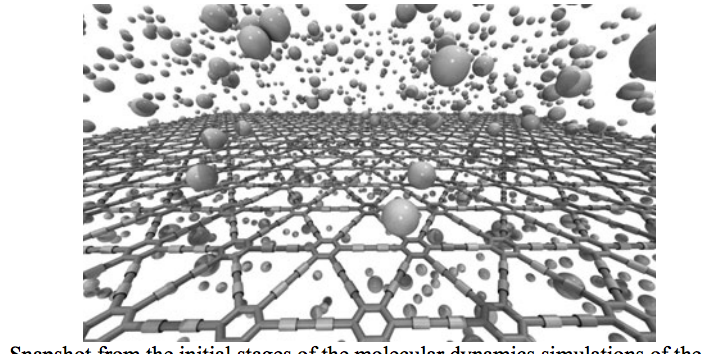
Autreto, PA; de Sousa, JM; Galvao, DS
On the Dynamics of Graphdiyne Hydrogenation Proceedings
Cambridge University Press, vol. 1549, 2013.
Resumo | Links | BibTeX | Tags: Graphdyine, Graphynes, Hydrogenation, Molecular Dynamics
@proceedings{autreto2013dynamics,
title = {On the Dynamics of Graphdiyne Hydrogenation},
author = {Autreto, PA and de Sousa, JM and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8915680&fileId=S1946427413006088},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1549},
pages = {59--64},
publisher = {Cambridge University Press},
abstract = {Graphene is a two-dimensional (2D) hexagonal array of carbon atoms in sp2-hybridized states. Graphene presents unique and exceptional electronic, thermal and mechanical properties. However, in its pristine state graphene is a gapless semiconductor, which poses some limitations to its use in some transistor electronics. Because of this there is a renewed interest in other possible two-dimensional carbon-based structures similar to graphene. Examples of this are graphynes and graphdiynes, which are two-dimensional structures, composed of carbon atoms in sp2 and sp-hybridized states. Graphdiynes (benzenoid rings connecting two acetylenic groups) were recently synthesized and they can be intrinsically nonzero gap systems. These systems can be easily hydrogenated and the amount of hydrogenation can be used to tune the band gap value. In this work we have investigated, through fully atomistic molecular dynamics simulations with reactive force field (ReaxFF), the structural and dynamics aspects of the hydrogenation mechanisms of graphdiyne membranes. Our results showed that depending on whether the atoms are in the benzenoid rings or as part of the acetylenic groups, the rates of hydrogenation are quite distinct and change in time in a very complex pattern. Initially, the most probable sites to be hydrogenated are the carbon atoms forming the triple bonds, as expected. But as the amount of hydrogenation increases in time this changes and then the carbon atoms forming single bonds become the preferential sites. The formation of correlated domains observed in hydrogenated graphene is no longer observed in the case of graphdiynes. We have also carried out ab initio DFT calculations for model structures in order to test the reliability of ReaxFF calculations.},
keywords = {Graphdyine, Graphynes, Hydrogenation, Molecular Dynamics},
pubstate = {published},
tppubtype = {proceedings}
}
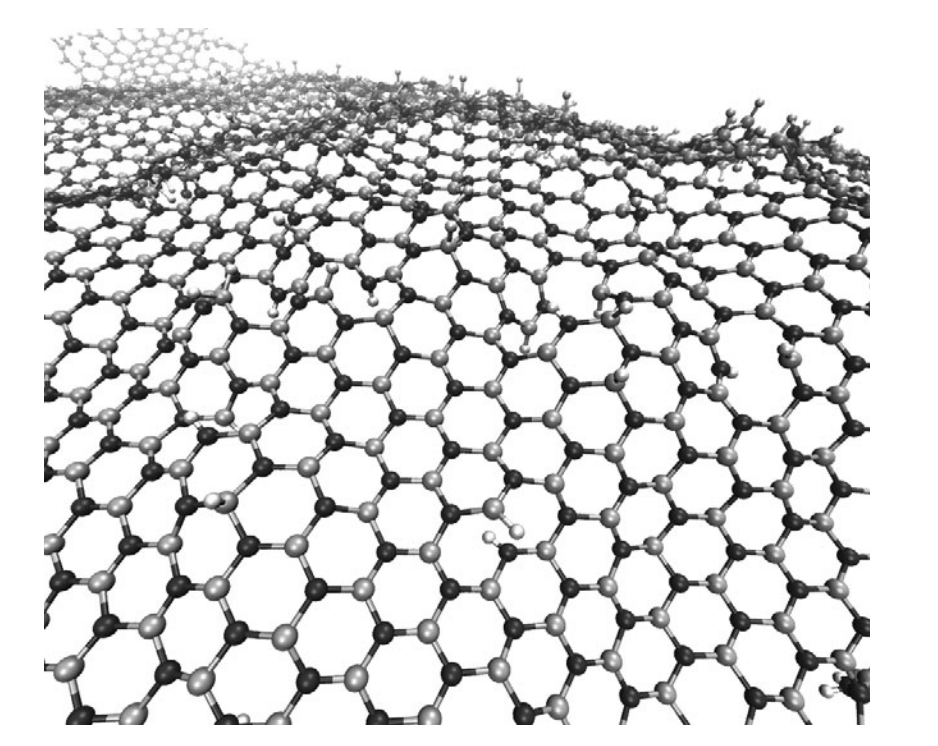
Perim, Eric; Paupitz, Ricardo; Autreto, PAS; Galvao, DS
The Hydrogenation Dynamics of h-BN Sheets Proceedings
Cambridge University Press, vol. 1549, 2013.
Resumo | Links | BibTeX | Tags: Boron Nitride, Hydrogenation, Molecular Dynamics, Nanotubes
@proceedings{perim2013hydrogenation,
title = {The Hydrogenation Dynamics of h-BN Sheets},
author = {Perim, Eric and Paupitz, Ricardo and Autreto, PAS and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8943477&fileId=S1946427413007938},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1549},
pages = {91--98},
publisher = {Cambridge University Press},
abstract = {Hexagonal boron nitride (h-BN), also known as white graphite, is the inorganic analogue of graphite. Single layers of both structures have been already experimentally realized.
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics of hydrogenation of h-BN single-layers membranes.
Our results show that the rate of hydrogenation atoms bonded to the membrane is highly dependent on the temperature and that only at low temperatures there is a preferential bond to boron atoms. Unlike graphanes (hydrogenated graphene), hydrogenated h-BN membranes do not exhibit the formation of correlated domains. Also, the out-of-plane deformations are more pronounced in comparison with the graphene case. After a critical number of incorporated hydrogen atoms the membrane become increasingly defective, lost its two-dimensional character and collapses. The hydrogen radial pair distribution and second-nearest neighbor correlations were also analyzed.},
keywords = {Boron Nitride, Hydrogenation, Molecular Dynamics, Nanotubes},
pubstate = {published},
tppubtype = {proceedings}
}
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics of hydrogenation of h-BN single-layers membranes.
Our results show that the rate of hydrogenation atoms bonded to the membrane is highly dependent on the temperature and that only at low temperatures there is a preferential bond to boron atoms. Unlike graphanes (hydrogenated graphene), hydrogenated h-BN membranes do not exhibit the formation of correlated domains. Also, the out-of-plane deformations are more pronounced in comparison with the graphene case. After a critical number of incorporated hydrogen atoms the membrane become increasingly defective, lost its two-dimensional character and collapses. The hydrogen radial pair distribution and second-nearest neighbor correlations were also analyzed.
2011
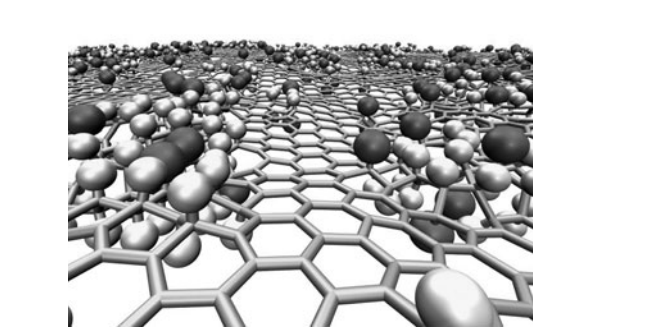
Autreto, Pedro AS; Flores, Marcelo Z; Legoas, Sergio B; Santos, Ricardo PB; Galvao, Douglas S
Cambridge University Press, vol. 1284, 2011.
Resumo | Links | BibTeX | Tags: Graphane, Graphene, Hydrogenation, Molecular Dynamics
@proceedings{autreto2011fully,
title = {A Fully Atomistic Reactive Molecular Dynamics Study on the Formation of Graphane from Graphene Hydrogenated Membranes.},
author = {Autreto, Pedro AS and Flores, Marcelo Z and Legoas, Sergio B and Santos, Ricardo PB and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8364784&fileId=S1946427411013583},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1284},
pages = {mrsf10--1284},
publisher = {Cambridge University Press},
abstract = {Using fully reactive molecular dynamics methodologies we investigated the structural and dynamical aspects of the fluorination mechanism leading to fluorographene formation from graphene membranes. Fluorination tends to produce significant defective areas on the membranes with variation on the typical carbon-carbon distances, sometimes with the presence of large holes due to carbon losses. The results obtained in our simulations are in good agreement with the broad distribution of values for the lattice parameter experimentally observed. We have also investigated mixed atmospheres composed by H and F atoms. When H is present in small quantities an expressive reduction on the rate of incorporation of fluorine was observed. On the other hand when fluorine atoms are present in small quantities in a hydrogen atmosphere, they induce an increasing on the hydrogen incorporation and the formation of locally self-organized structure of adsorbed H and F atoms.},
keywords = {Graphane, Graphene, Hydrogenation, Molecular Dynamics},
pubstate = {published},
tppubtype = {proceedings}
}
2009
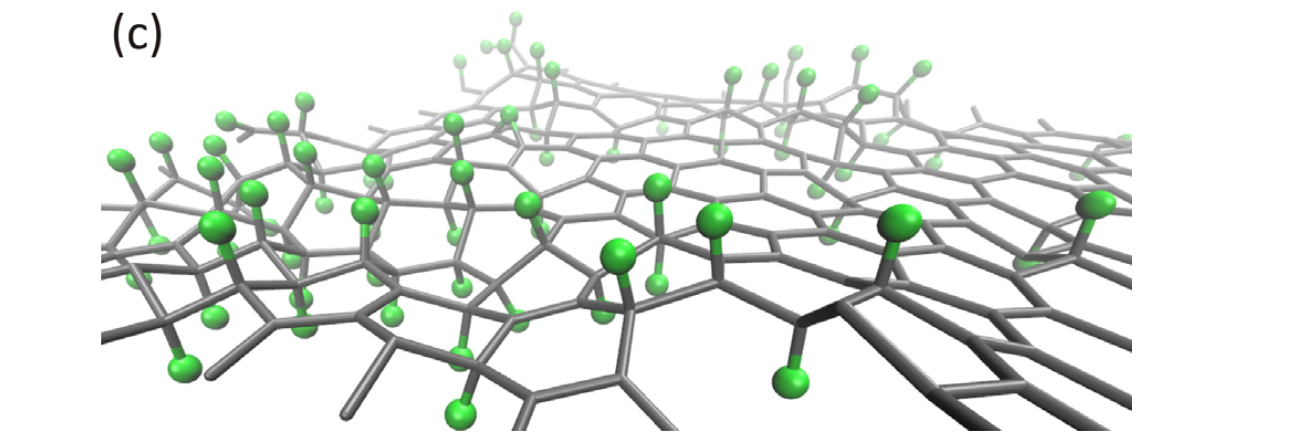
Flores, Marcelo ZS; Autreto, Pedro AS; Legoas, Sergio B; Galvao, Douglas S
Graphene to graphane: a theoretical study Journal Article
Em: Nanotechnology, vol. 20, não 46, pp. 465704, 2009.
Resumo | Links | BibTeX | Tags: Functionalization, Graphanes, Graphene, Hydrogenation
@article{flores2009graphene,
title = {Graphene to graphane: a theoretical study},
author = {Flores, Marcelo ZS and Autreto, Pedro AS and Legoas, Sergio B and Galvao, Douglas S},
url = {http://iopscience.iop.org/0957-4484/20/46/465704},
year = {2009},
date = {2009-01-01},
journal = {Nanotechnology},
volume = {20},
number = {46},
pages = {465704},
publisher = {IOP Publishing},
abstract = {Graphane is a two-dimensional system consisting of a single layer of fully saturated (sp3 hybridization) carbon atoms. In an ideal graphane structure C–H bonds exhibit an alternating pattern (up and down with relation to the plane defined by the carbon atoms). In this work we have investigated, using ab initio and reactive molecular dynamics simulations, the role of H frustration (breaking the H atoms' up and down alternating pattern) in graphane-like structures. Our results show that a significant percentage of uncorrelated H frustrated domains are formed in the early stages of the hydrogenation process leading to membrane shrinkage and extensive membrane corrugations. These results also suggest that large domains of perfect graphane-like structures are unlikely to be formed, as H frustrated domains are always present.
},
keywords = {Functionalization, Graphanes, Graphene, Hydrogenation},
pubstate = {published},
tppubtype = {article}
}
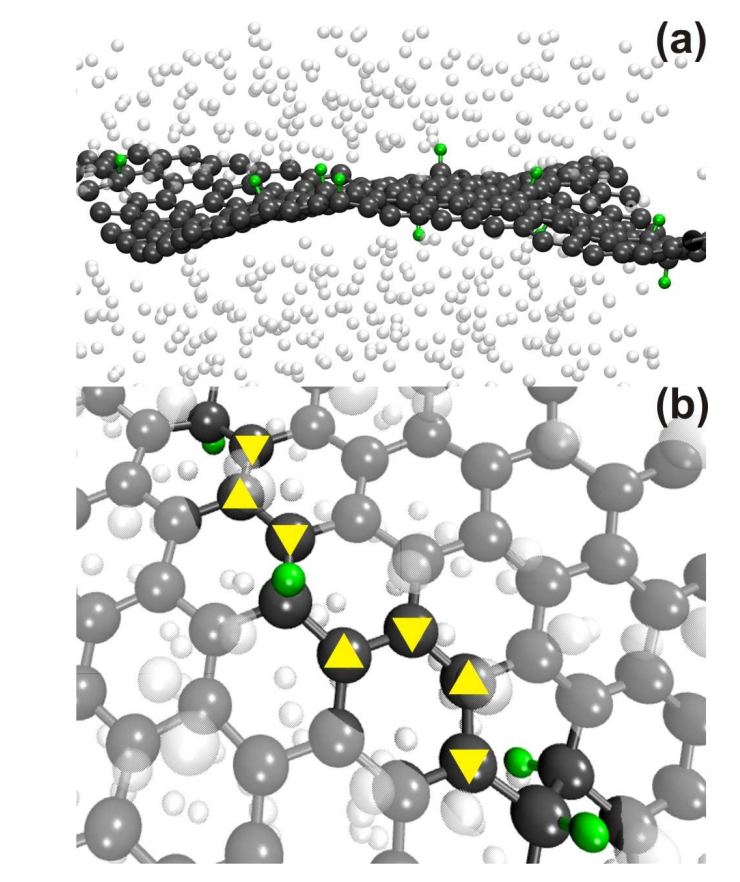
Legoas, Sergio B; Autreto, Pedro AS; Flores, Marcelo ZS; Galvao, Douglas S
Graphene to graphane: the role of H frustration in lattice contraction Journal Article
Em: arXiv preprint arXiv:0903.0278, 2009.
Resumo | Links | BibTeX | Tags: Functionalization, Graphane, Graphene, Hydrogenation
@article{legoas2009graphene,
title = {Graphene to graphane: the role of H frustration in lattice contraction},
author = {Legoas, Sergio B and Autreto, Pedro AS and Flores, Marcelo ZS and Galvao, Douglas S},
url = {http://arxiv.org/abs/0903.0278},
year = {2009},
date = {2009-01-01},
journal = {arXiv preprint arXiv:0903.0278},
abstract = {Graphane is a two-dimensional system consisting of a single planar layer of fully saturated (sp3 hybridization) carbon atoms with H atoms attached to them in an alternating pattern (up and down with relation to the plane defined by the carbon atoms). Stable graphane structures were theoretically predicted to exist some years ago and just experimentally realized through hydrogenation of graphene membranes. In this work we have investigated using textit{ab initio} and reactive molecular dynamics the role of H frustration (breaking the H atoms up and down alternating pattern) in graphane-like structures. Our results show that H frustration significantly contributes to lattice contraction. The dynamical aspects of converting graphene to graphane is also addressed.},
keywords = {Functionalization, Graphane, Graphene, Hydrogenation},
pubstate = {published},
tppubtype = {article}
}

