
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ
Brunetto, Gustavo; Sato, Fernando; Bouju, Xavier; Galvao, Douglas S
The First Molecular Wheel: A Theoretical Investigation Proceedings
Cambridge University Press, vol. 1286, 2011.
@proceedings{brunetto2011first,
title = {The First Molecular Wheel: A Theoretical Investigation},
author = {Brunetto, Gustavo and Sato, Fernando and Bouju, Xavier and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=7975705&fileId=S1946427411000133},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1286},
pages = {mrsf10--1286},
publisher = {Cambridge University Press},
abstract = {Recently, the first molecular nanowheel was synthesized and characterized from Scanning Tunneling Microscope (STM) experiments. It was demonstrated that a specifically designed hydrocarbon molecule (C44H24) could roll on a copper substrate along the [110] surface direction. In this work we report a preliminary theoretical analysis of the isolated molecule and of its rolling processes on different Cu surfaces. We have used ab initio and classical molecular dynamics methods. The simulations showed that the rolling mechanism is only possible for the [110] surface. In this case, the spatial separation among rows of copper atoms is enough to ‘trap’ the molecule and to create the necessary torque to roll it. Other surface orientations ([111] and [100]) are too smooth and cannot provide the necessary torque for the rolling process.},
keywords = {},
pubstate = {published},
tppubtype = {proceedings}
}
Sato, F; Legoas, SB; Otero, R; Hummelink, F; Thostrup, P; Lægsgaard, E; Stensgaard, I; Besenbacher, F; Galvao, DS
Adsorption configuration effects on the surface diffusion of large organic molecules: The case of Violet Lander Journal Article
Em: The Journal of chemical physics, vol. 133, não 22, pp. 224702, 2010.
@article{sato2010adsorption,
title = {Adsorption configuration effects on the surface diffusion of large organic molecules: The case of Violet Lander},
author = {Sato, F and Legoas, SB and Otero, R and Hummelink, F and Thostrup, P and Lægsgaard, E and Stensgaard, I and Besenbacher, F and Galvao, DS},
url = {http://scitation.aip.org/content/aip/journal/jcp/133/22/10.1063/1.3512623},
year = {2010},
date = {2010-01-01},
journal = {The Journal of chemical physics},
volume = {133},
number = {22},
pages = {224702},
publisher = {AIP Publishing},
abstract = {Violet Lander (C108H104) is a large organic molecule that when deposited on Cu(110) surface exhibits lock-and-key like behavior [Otero et al., Nature Mater. 3, 779 (2004)]. In this work, we report a detailed fully atomistic molecular mechanics and molecular dynamics study of this phenomenon. Our results show that it has its physical basis on the interplay of the molecular hydrogens and the Cu(110) atomic spacing, which is a direct consequence of the matching between molecule and surface dimensions. This information could be used to find new molecules capable of displaying lock-and-key behavior with new potential applications in nanotechnology.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Otero, Roberto; Hummelink, Frauke; Sato, Fernando; Legoas, Sergio B; Thostrup, Peter; Lægsgaard, Erik; Stensgaard, Ivan; Galvao, Douglas S; Besenbacher, Flemming
Lock-and-key effect in the surface diffusion of large organic molecules probed by STM Journal Article
Em: Nature Materials, vol. 3, não 11, pp. 779–782, 2004.
@article{otero2004lock,
title = {Lock-and-key effect in the surface diffusion of large organic molecules probed by STM},
author = {Otero, Roberto and Hummelink, Frauke and Sato, Fernando and Legoas, Sergio B and Thostrup, Peter and Lægsgaard, Erik and Stensgaard, Ivan and Galvao, Douglas S and Besenbacher, Flemming},
url = {http://www.nature.com/nmat/journal/v3/n11/full/nmat1243.html},
year = {2004},
date = {2004-01-01},
journal = {Nature Materials},
volume = {3},
number = {11},
pages = {779--782},
publisher = {Nature Publishing Group},
abstract = {A nanoscale understanding of the complex dynamics of large molecules at surfaces is essential for the bottom-up design of molecular nanostructures1, 2, 3, 4, 5, 6, 7, 8. Here we show that we can change the diffusion coefficient of the complex organic molecule known as Violet Lander (VL, C108H104) on Cu(110) by two orders of magnitude by using the STM at low temperatures to switch between two adsorption configurations that differ only in the molecular orientation with respect to the substrate lattice. From an interplay with molecular dynamics simulations, we interpret the results within a lock-and-key model similar to the one driving the recognition between biomolecules: the molecule (key) is immobilized only when its orientation is such that the molecular shape fits the atomic lattice of the surface (lock); otherwise the molecule is highly mobile.
Introduction
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Introduction
Galvao, DS; Braga, SF; Barone, PMVB; Dantas, SO
Dual-Mode Optical Molecular Switching Systems for Organic Memories Proceedings
Cambridge Univ Press, vol. 708, 2002.
@proceedings{galvao2002dual,
title = {Dual-Mode Optical Molecular Switching Systems for Organic Memories},
author = {Galvao, DS and Braga, SF and Barone, PMVB and Dantas, SO},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8214320},
year = {2002},
date = {2002-01-01},
journal = {MATERIALS RESEARCH SOCIETY SYMPOSIUM PROCEEDINGS},
volume = {708},
pages = {335--340},
publisher = {Cambridge Univ Press},
abstract = {The synthesis of dual-mode optical molecular switching systems has been recently achieved. These systems were based on chiral helical-shaped alkenes in which the chirality can be reversibly modulated by light. In this work we report a theoretical study on the geometric and spectroscopic properties of these structures using the well-known semi-empirical methods PM3 (Parametric Method 3) and ZINDO/S-CI (Zerner's Intermediate Neglect of Differential Overlap -Spectroscopic - Configuration Interaction). Our results show that there are two stable conformers very close in energy for each possible molecular helicity presenting a barrier of ∼40 kcal/mol for bond rotation along the main molecular axis. Under protonation these barriers increase significantly and might explain why the protonation leads to the blocking of the switching process. We propose a scheme for the switching mechanism based on charge transfer and conformational changes during the isomer interconversion.},
keywords = {},
pubstate = {published},
tppubtype = {proceedings}
}
2011
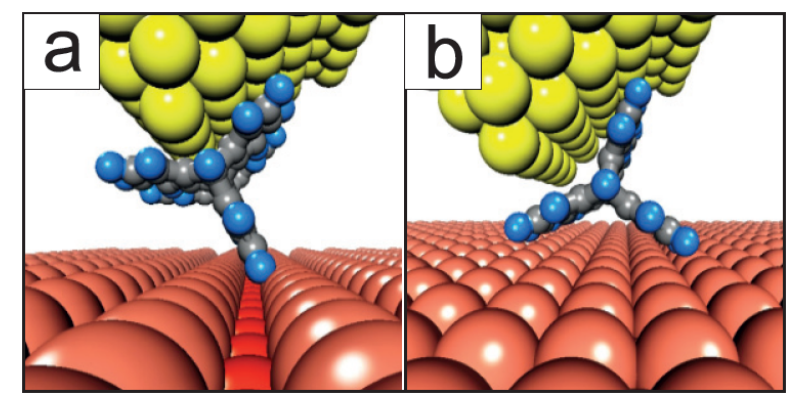
Brunetto, Gustavo; Sato, Fernando; Bouju, Xavier; Galvao, Douglas S
The First Molecular Wheel: A Theoretical Investigation Proceedings
Cambridge University Press, vol. 1286, 2011.
Resumo | Links | BibTeX | Tags: Molecular Dynamics, Molecular Electronics, Nanowheel
@proceedings{brunetto2011first,
title = {The First Molecular Wheel: A Theoretical Investigation},
author = {Brunetto, Gustavo and Sato, Fernando and Bouju, Xavier and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=7975705&fileId=S1946427411000133},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1286},
pages = {mrsf10--1286},
publisher = {Cambridge University Press},
abstract = {Recently, the first molecular nanowheel was synthesized and characterized from Scanning Tunneling Microscope (STM) experiments. It was demonstrated that a specifically designed hydrocarbon molecule (C44H24) could roll on a copper substrate along the [110] surface direction. In this work we report a preliminary theoretical analysis of the isolated molecule and of its rolling processes on different Cu surfaces. We have used ab initio and classical molecular dynamics methods. The simulations showed that the rolling mechanism is only possible for the [110] surface. In this case, the spatial separation among rows of copper atoms is enough to ‘trap’ the molecule and to create the necessary torque to roll it. Other surface orientations ([111] and [100]) are too smooth and cannot provide the necessary torque for the rolling process.},
keywords = {Molecular Dynamics, Molecular Electronics, Nanowheel},
pubstate = {published},
tppubtype = {proceedings}
}
2010
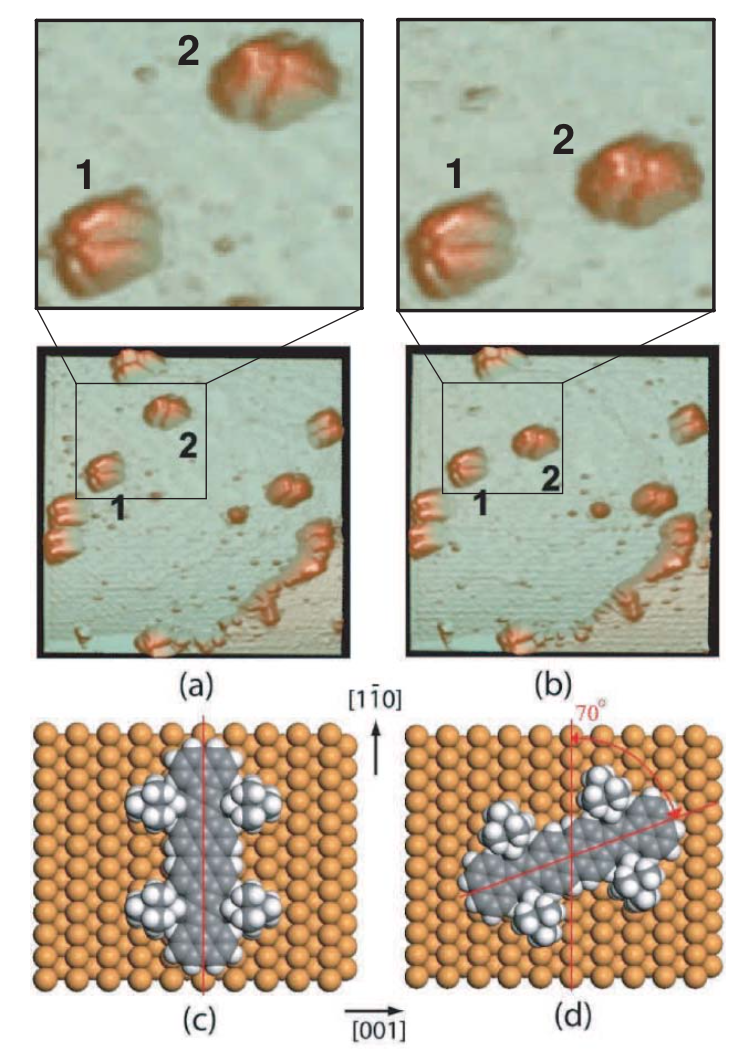
Sato, F; Legoas, SB; Otero, R; Hummelink, F; Thostrup, P; Lægsgaard, E; Stensgaard, I; Besenbacher, F; Galvao, DS
Adsorption configuration effects on the surface diffusion of large organic molecules: The case of Violet Lander Journal Article
Em: The Journal of chemical physics, vol. 133, não 22, pp. 224702, 2010.
Resumo | Links | BibTeX | Tags: DFT, Diffusion, Molecular Electronics, STM, Violet Lander
@article{sato2010adsorption,
title = {Adsorption configuration effects on the surface diffusion of large organic molecules: The case of Violet Lander},
author = {Sato, F and Legoas, SB and Otero, R and Hummelink, F and Thostrup, P and Lægsgaard, E and Stensgaard, I and Besenbacher, F and Galvao, DS},
url = {http://scitation.aip.org/content/aip/journal/jcp/133/22/10.1063/1.3512623},
year = {2010},
date = {2010-01-01},
journal = {The Journal of chemical physics},
volume = {133},
number = {22},
pages = {224702},
publisher = {AIP Publishing},
abstract = {Violet Lander (C108H104) is a large organic molecule that when deposited on Cu(110) surface exhibits lock-and-key like behavior [Otero et al., Nature Mater. 3, 779 (2004)]. In this work, we report a detailed fully atomistic molecular mechanics and molecular dynamics study of this phenomenon. Our results show that it has its physical basis on the interplay of the molecular hydrogens and the Cu(110) atomic spacing, which is a direct consequence of the matching between molecule and surface dimensions. This information could be used to find new molecules capable of displaying lock-and-key behavior with new potential applications in nanotechnology.},
keywords = {DFT, Diffusion, Molecular Electronics, STM, Violet Lander},
pubstate = {published},
tppubtype = {article}
}
2004
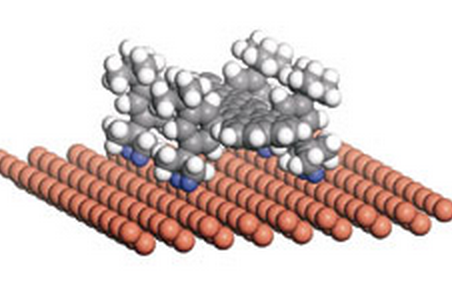
Otero, Roberto; Hummelink, Frauke; Sato, Fernando; Legoas, Sergio B; Thostrup, Peter; Lægsgaard, Erik; Stensgaard, Ivan; Galvao, Douglas S; Besenbacher, Flemming
Lock-and-key effect in the surface diffusion of large organic molecules probed by STM Journal Article
Em: Nature Materials, vol. 3, não 11, pp. 779–782, 2004.
Resumo | Links | BibTeX | Tags: Landers, Molecular Dynamics, Molecular Electronics, STM, top20
@article{otero2004lock,
title = {Lock-and-key effect in the surface diffusion of large organic molecules probed by STM},
author = {Otero, Roberto and Hummelink, Frauke and Sato, Fernando and Legoas, Sergio B and Thostrup, Peter and Lægsgaard, Erik and Stensgaard, Ivan and Galvao, Douglas S and Besenbacher, Flemming},
url = {http://www.nature.com/nmat/journal/v3/n11/full/nmat1243.html},
year = {2004},
date = {2004-01-01},
journal = {Nature Materials},
volume = {3},
number = {11},
pages = {779--782},
publisher = {Nature Publishing Group},
abstract = {A nanoscale understanding of the complex dynamics of large molecules at surfaces is essential for the bottom-up design of molecular nanostructures1, 2, 3, 4, 5, 6, 7, 8. Here we show that we can change the diffusion coefficient of the complex organic molecule known as Violet Lander (VL, C108H104) on Cu(110) by two orders of magnitude by using the STM at low temperatures to switch between two adsorption configurations that differ only in the molecular orientation with respect to the substrate lattice. From an interplay with molecular dynamics simulations, we interpret the results within a lock-and-key model similar to the one driving the recognition between biomolecules: the molecule (key) is immobilized only when its orientation is such that the molecular shape fits the atomic lattice of the surface (lock); otherwise the molecule is highly mobile.
Introduction
},
keywords = {Landers, Molecular Dynamics, Molecular Electronics, STM, top20},
pubstate = {published},
tppubtype = {article}
}
Introduction
2002
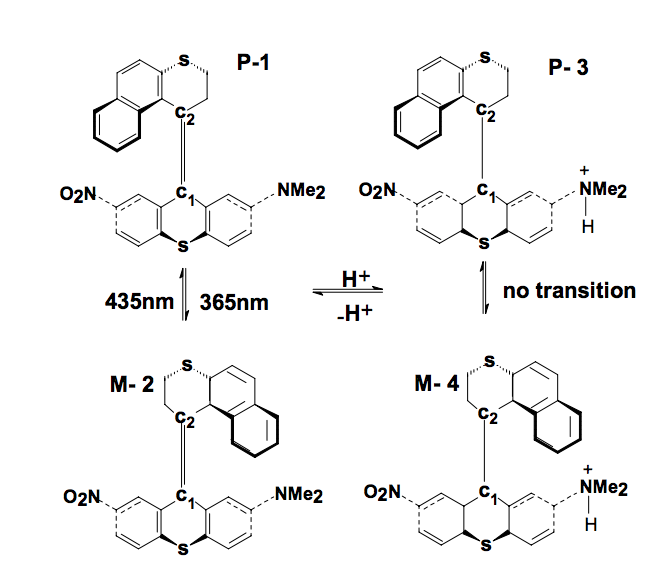
Galvao, DS; Braga, SF; Barone, PMVB; Dantas, SO
Dual-Mode Optical Molecular Switching Systems for Organic Memories Proceedings
Cambridge Univ Press, vol. 708, 2002.
Resumo | Links | BibTeX | Tags: Electronic Structure, Molecular Electronics
@proceedings{galvao2002dual,
title = {Dual-Mode Optical Molecular Switching Systems for Organic Memories},
author = {Galvao, DS and Braga, SF and Barone, PMVB and Dantas, SO},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8214320},
year = {2002},
date = {2002-01-01},
journal = {MATERIALS RESEARCH SOCIETY SYMPOSIUM PROCEEDINGS},
volume = {708},
pages = {335--340},
publisher = {Cambridge Univ Press},
abstract = {The synthesis of dual-mode optical molecular switching systems has been recently achieved. These systems were based on chiral helical-shaped alkenes in which the chirality can be reversibly modulated by light. In this work we report a theoretical study on the geometric and spectroscopic properties of these structures using the well-known semi-empirical methods PM3 (Parametric Method 3) and ZINDO/S-CI (Zerner's Intermediate Neglect of Differential Overlap -Spectroscopic - Configuration Interaction). Our results show that there are two stable conformers very close in energy for each possible molecular helicity presenting a barrier of ∼40 kcal/mol for bond rotation along the main molecular axis. Under protonation these barriers increase significantly and might explain why the protonation leads to the blocking of the switching process. We propose a scheme for the switching mechanism based on charge transfer and conformational changes during the isomer interconversion.},
keywords = {Electronic Structure, Molecular Electronics},
pubstate = {published},
tppubtype = {proceedings}
}

