
Fonseca, AF; Dantas, SO; Galvao, DS; Zhang, D; Sinnott, SB
The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review) Journal Article
In: 2019.
@article{Fonseca2019d,
title = {The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review)},
author = {Fonseca, AF and Dantas, SO and Galvao, DS and Zhang, D and Sinnott, SB},
year = {2019},
date = {2019-04-03},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
AF; Dantas Fonseca, SO; Galvao
The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study Online
2019, (ArXiv preprint).
@online{Fonseca2019b,
title = {The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study},
author = {Fonseca, AF; Dantas, SO; Galvao, DS; Zhang, D; Sinnott SB},
url = {https://arxiv.org/pdf/1904.09871.pdf},
year = {2019},
date = {2019-03-22},
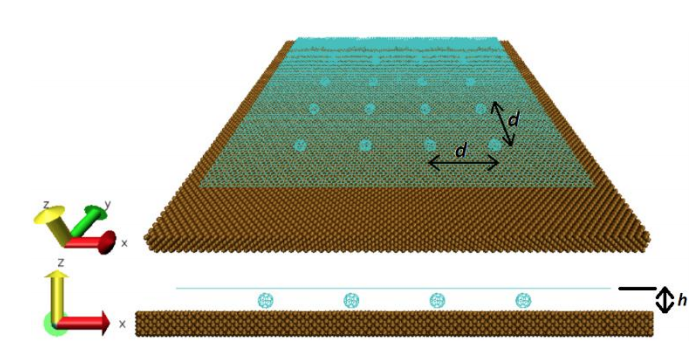
abstract = {Two experimental studies reported the spontaneous formation of amorphous and crystalline
structures of C60 intercalated between graphene and a substrate. They observed interesting
phenomena ranging from reaction between C60 molecules under graphene to graphene
sagging between the molecules and control of strain in graphene. Motivated by these works,
we performed fully atomistic reactive molecular dynamics simulations to study the formation
and thermal stability of graphene wrinkles as well as graphene attachment to and detachment
from the substrate when graphene is laid over a previously distributed array of C60 molecules
on a copper substrate at different values of temperature. As graphene compresses the C60
molecules against the substrate, and graphene attachment to the substrate between C60s
(“C60S” stands for plural of C60) depends on the height of graphene wrinkles, configurations
with both frozen and non-frozen C60s structures were investigated in order to verify the
experimental result of stable sagged graphene when the distance between C60s is about 4 nm
and height of graphene wrinkles is about 0.8 nm. Below the distance of 4 nm between C60s,
graphene becomes locally suspended and less strained. We show that this happens when C60s
are allowed to deform under the compressive action of graphene. If we keep the C60s frozen,
spontaneous “blanketing” of graphene happens only when the distance between them are
equal or above 7 nm. Both above results for the existence of stable sagged graphene for C60
distances of 4 or 7 nm are shown to agree with a mechanical model relating the rigidity of
graphene to the energy of graphene-substrate adhesion. Although the studies of intercalation
of molecules on interfaces formed by graphene-substrate are motivated by finding out ways to
control wrinkling and strain in graphene, our work reveals the shape and structure of
intercalated molecules and the role of stability and wrinkling on final structure of graphene.
In particular, this study might help the development of 2D confined nanoreactors that are
considered in literature to be the next advanced step on chemical reactions.},
note = {ArXiv preprint},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
structures of C60 intercalated between graphene and a substrate. They observed interesting
phenomena ranging from reaction between C60 molecules under graphene to graphene
sagging between the molecules and control of strain in graphene. Motivated by these works,
we performed fully atomistic reactive molecular dynamics simulations to study the formation
and thermal stability of graphene wrinkles as well as graphene attachment to and detachment
from the substrate when graphene is laid over a previously distributed array of C60 molecules
on a copper substrate at different values of temperature. As graphene compresses the C60
molecules against the substrate, and graphene attachment to the substrate between C60s
(“C60S” stands for plural of C60) depends on the height of graphene wrinkles, configurations
with both frozen and non-frozen C60s structures were investigated in order to verify the
experimental result of stable sagged graphene when the distance between C60s is about 4 nm
and height of graphene wrinkles is about 0.8 nm. Below the distance of 4 nm between C60s,
graphene becomes locally suspended and less strained. We show that this happens when C60s
are allowed to deform under the compressive action of graphene. If we keep the C60s frozen,
spontaneous “blanketing” of graphene happens only when the distance between them are
equal or above 7 nm. Both above results for the existence of stable sagged graphene for C60
distances of 4 or 7 nm are shown to agree with a mechanical model relating the rigidity of
graphene to the energy of graphene-substrate adhesion. Although the studies of intercalation
of molecules on interfaces formed by graphene-substrate are motivated by finding out ways to
control wrinkling and strain in graphene, our work reveals the shape and structure of
intercalated molecules and the role of stability and wrinkling on final structure of graphene.
In particular, this study might help the development of 2D confined nanoreactors that are
considered in literature to be the next advanced step on chemical reactions.
AF; Dantas Fonseca, SO; Galvao
The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review) Journal Article
In: 2019.
@article{Fonseca2019c,
title = {The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review)},
author = {Fonseca, AF; Dantas, SO; Galvao, DS; Zhang, D; Sinnott SB},
year = {2019},
date = {2019-03-15},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Coluci, Vitor R; Sato, Fernando; Braga, Scheila F; Skaf, Munir S; Galvao, Douglas S
Rotational dynamics and polymerization of C60 in C60-cubane crystals: A molecular dynamics study Journal Article
In: The Journal of Chemical Physics, vol. 129, no. 6, pp. 064506, 2008.
@article{coluci2008rotational,
title = {Rotational dynamics and polymerization of C60 in C60-cubane crystals: A molecular dynamics study},
author = {Coluci, Vitor R and Sato, Fernando and Braga, Scheila F and Skaf, Munir S and Galvao, Douglas S},
url = {http://scitation.aip.org/content/aip/journal/jcp/129/6/10.1063/1.2965885},
year = {2008},
date = {2008-01-01},
journal = {The Journal of Chemical Physics},
volume = {129},
number = {6},
pages = {064506},
publisher = {AIP Publishing},
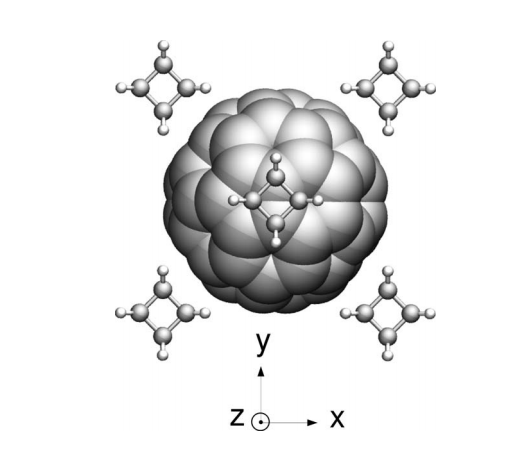
abstract = {We report classical and tight-binding molecular dynamics simulations of the C60fullerene and cubane molecular crystal in order to investigate the intermolecular dynamics and polymerization processes. Our results show that, for 200 and 400 K, cubane molecules remain basically fixed, presenting only thermal vibrations, while C60fullerenes show rotational motions. Fullerenes perform “free” rotational motions at short times (≲1 ps), small amplitude hindered rotational motions (librations) at intermediate times, and rotational diffusive dynamics at long times (≳10 ps). The mechanisms underlying these dynamics are presented. Random copolymerizations among cubanes and fullerenes were observed when temperature is increased, leading to the formation of a disordered structure. Changes in the radial distribution function and electronic density of states indicate the coexistence of amorphous and crystalline phases. The different conformational phases that cubanes and fullerenes undergo during the copolymerization process are discussed.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Legoas, Sergio B; Giro, Ronaldo; Galvao, Douglas S
Molecular dynamics simulations of C6) nanobearings Journal Article
In: Chemical physics letters, vol. 386, no. 4, pp. 425–429, 2004.
@article{legoas2004molecular,
title = {Molecular dynamics simulations of C6) nanobearings},
author = {Legoas, Sergio B and Giro, Ronaldo and Galvao, Douglas S},
url = {http://www.sciencedirect.com/science/article/pii/S000926140400168X},
year = {2004},
date = {2004-01-01},
journal = {Chemical physics letters},
volume = {386},
number = {4},
pages = {425--429},
publisher = {Elsevier},
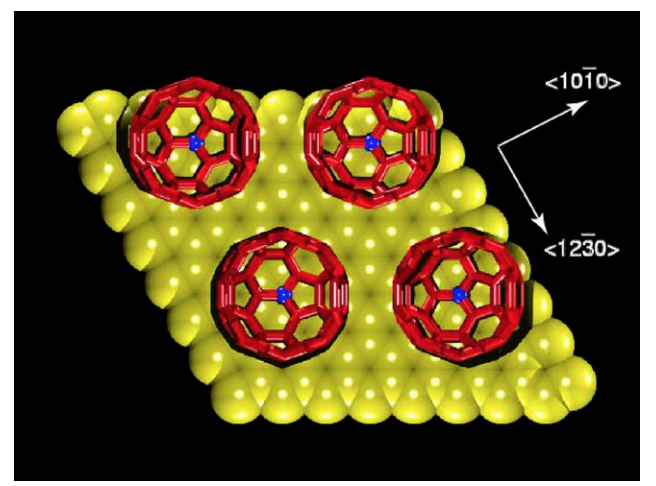
abstract = {Recently was reported an ultra-lubricated system based on C60 molecules deposited over graphite layers. In that work a stick-slip rolling model for C60 molecules was proposed to explain the observed ultra-low friction force. In this Letter, we report the first molecular dynamics studies for these systems. Our results show that the AB stacking is not observed and the main experimental features can be explained without invoking stick-slip motions.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2019
Fonseca, AF; Dantas, SO; Galvao, DS; Zhang, D; Sinnott, SB
The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review) Journal Article
In: 2019.
BibTeX | Tags: C60, Graphene, Molecular Dynamics
@article{Fonseca2019d,
title = {The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review)},
author = {Fonseca, AF and Dantas, SO and Galvao, DS and Zhang, D and Sinnott, SB},
year = {2019},
date = {2019-04-03},
keywords = {C60, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
AF; Dantas Fonseca, SO; Galvao
The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study Online
2019, (ArXiv preprint).
Abstract | Links | BibTeX | Tags: C60, Graphene, Molecular Dynamics
@online{Fonseca2019b,
title = {The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study},
author = {Fonseca, AF; Dantas, SO; Galvao, DS; Zhang, D; Sinnott SB},
url = {https://arxiv.org/pdf/1904.09871.pdf},
year = {2019},
date = {2019-03-22},
abstract = {Two experimental studies reported the spontaneous formation of amorphous and crystalline
structures of C60 intercalated between graphene and a substrate. They observed interesting
phenomena ranging from reaction between C60 molecules under graphene to graphene
sagging between the molecules and control of strain in graphene. Motivated by these works,
we performed fully atomistic reactive molecular dynamics simulations to study the formation
and thermal stability of graphene wrinkles as well as graphene attachment to and detachment
from the substrate when graphene is laid over a previously distributed array of C60 molecules
on a copper substrate at different values of temperature. As graphene compresses the C60
molecules against the substrate, and graphene attachment to the substrate between C60s
(“C60S” stands for plural of C60) depends on the height of graphene wrinkles, configurations
with both frozen and non-frozen C60s structures were investigated in order to verify the
experimental result of stable sagged graphene when the distance between C60s is about 4 nm
and height of graphene wrinkles is about 0.8 nm. Below the distance of 4 nm between C60s,
graphene becomes locally suspended and less strained. We show that this happens when C60s
are allowed to deform under the compressive action of graphene. If we keep the C60s frozen,
spontaneous “blanketing” of graphene happens only when the distance between them are
equal or above 7 nm. Both above results for the existence of stable sagged graphene for C60
distances of 4 or 7 nm are shown to agree with a mechanical model relating the rigidity of
graphene to the energy of graphene-substrate adhesion. Although the studies of intercalation
of molecules on interfaces formed by graphene-substrate are motivated by finding out ways to
control wrinkling and strain in graphene, our work reveals the shape and structure of
intercalated molecules and the role of stability and wrinkling on final structure of graphene.
In particular, this study might help the development of 2D confined nanoreactors that are
considered in literature to be the next advanced step on chemical reactions.},
note = {ArXiv preprint},
keywords = {C60, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}
structures of C60 intercalated between graphene and a substrate. They observed interesting
phenomena ranging from reaction between C60 molecules under graphene to graphene
sagging between the molecules and control of strain in graphene. Motivated by these works,
we performed fully atomistic reactive molecular dynamics simulations to study the formation
and thermal stability of graphene wrinkles as well as graphene attachment to and detachment
from the substrate when graphene is laid over a previously distributed array of C60 molecules
on a copper substrate at different values of temperature. As graphene compresses the C60
molecules against the substrate, and graphene attachment to the substrate between C60s
(“C60S” stands for plural of C60) depends on the height of graphene wrinkles, configurations
with both frozen and non-frozen C60s structures were investigated in order to verify the
experimental result of stable sagged graphene when the distance between C60s is about 4 nm
and height of graphene wrinkles is about 0.8 nm. Below the distance of 4 nm between C60s,
graphene becomes locally suspended and less strained. We show that this happens when C60s
are allowed to deform under the compressive action of graphene. If we keep the C60s frozen,
spontaneous “blanketing” of graphene happens only when the distance between them are
equal or above 7 nm. Both above results for the existence of stable sagged graphene for C60
distances of 4 or 7 nm are shown to agree with a mechanical model relating the rigidity of
graphene to the energy of graphene-substrate adhesion. Although the studies of intercalation
of molecules on interfaces formed by graphene-substrate are motivated by finding out ways to
control wrinkling and strain in graphene, our work reveals the shape and structure of
intercalated molecules and the role of stability and wrinkling on final structure of graphene.
In particular, this study might help the development of 2D confined nanoreactors that are
considered in literature to be the next advanced step on chemical reactions.
AF; Dantas Fonseca, SO; Galvao
The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review) Journal Article
In: 2019.
BibTeX | Tags: C60, Graphene, Molecular Dynamics
@article{Fonseca2019c,
title = {The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review)},
author = {Fonseca, AF; Dantas, SO; Galvao, DS; Zhang, D; Sinnott SB},
year = {2019},
date = {2019-03-15},
keywords = {C60, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
2008
Coluci, Vitor R; Sato, Fernando; Braga, Scheila F; Skaf, Munir S; Galvao, Douglas S
Rotational dynamics and polymerization of C60 in C60-cubane crystals: A molecular dynamics study Journal Article
In: The Journal of Chemical Physics, vol. 129, no. 6, pp. 064506, 2008.
Abstract | Links | BibTeX | Tags: C60, C70, Cubanes, Fullerenes, Molecular Dynamics, Rotor-Stator
@article{coluci2008rotational,
title = {Rotational dynamics and polymerization of C60 in C60-cubane crystals: A molecular dynamics study},
author = {Coluci, Vitor R and Sato, Fernando and Braga, Scheila F and Skaf, Munir S and Galvao, Douglas S},
url = {http://scitation.aip.org/content/aip/journal/jcp/129/6/10.1063/1.2965885},
year = {2008},
date = {2008-01-01},
journal = {The Journal of Chemical Physics},
volume = {129},
number = {6},
pages = {064506},
publisher = {AIP Publishing},
abstract = {We report classical and tight-binding molecular dynamics simulations of the C60fullerene and cubane molecular crystal in order to investigate the intermolecular dynamics and polymerization processes. Our results show that, for 200 and 400 K, cubane molecules remain basically fixed, presenting only thermal vibrations, while C60fullerenes show rotational motions. Fullerenes perform “free” rotational motions at short times (≲1 ps), small amplitude hindered rotational motions (librations) at intermediate times, and rotational diffusive dynamics at long times (≳10 ps). The mechanisms underlying these dynamics are presented. Random copolymerizations among cubanes and fullerenes were observed when temperature is increased, leading to the formation of a disordered structure. Changes in the radial distribution function and electronic density of states indicate the coexistence of amorphous and crystalline phases. The different conformational phases that cubanes and fullerenes undergo during the copolymerization process are discussed.},
keywords = {C60, C70, Cubanes, Fullerenes, Molecular Dynamics, Rotor-Stator},
pubstate = {published},
tppubtype = {article}
}
2004
Legoas, Sergio B; Giro, Ronaldo; Galvao, Douglas S
Molecular dynamics simulations of C6) nanobearings Journal Article
In: Chemical physics letters, vol. 386, no. 4, pp. 425–429, 2004.
Abstract | Links | BibTeX | Tags: C60, Fullerenes, Molecular Dynamics, Nanobearing, Tribology
@article{legoas2004molecular,
title = {Molecular dynamics simulations of C6) nanobearings},
author = {Legoas, Sergio B and Giro, Ronaldo and Galvao, Douglas S},
url = {http://www.sciencedirect.com/science/article/pii/S000926140400168X},
year = {2004},
date = {2004-01-01},
journal = {Chemical physics letters},
volume = {386},
number = {4},
pages = {425--429},
publisher = {Elsevier},
abstract = {Recently was reported an ultra-lubricated system based on C60 molecules deposited over graphite layers. In that work a stick-slip rolling model for C60 molecules was proposed to explain the observed ultra-low friction force. In this Letter, we report the first molecular dynamics studies for these systems. Our results show that the AB stacking is not observed and the main experimental features can be explained without invoking stick-slip motions.
},
keywords = {C60, Fullerenes, Molecular Dynamics, Nanobearing, Tribology},
pubstate = {published},
tppubtype = {article}
}
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ

