
Bizao, Rafael A; Botari, Tiago; Perim, Eric; Pugno, Nicola M; Galvao, Douglas S
Mechanical properties and fracture patterns of graphene (graphitic) nanowiggles Journal Article
In: Carbon, vol. 119, pp. 431-437, 2017, (See also ArxIv version: https://arxiv.org/abs/1702.01100).
@article{Bizao2017b,
title = {Mechanical properties and fracture patterns of graphene (graphitic) nanowiggles},
author = {Bizao, Rafael A and Botari, Tiago and Perim, Eric and Pugno, Nicola M and Galvao, Douglas S},
url = {http://www.sciencedirect.com/science/article/pii/S0008622317303743},
doi = {10.1016/j.carbon.2017.04.018},
year = {2017},
date = {2017-04-14},
journal = {Carbon},
volume = {119},
pages = {431-437},
abstract = {Graphene nanowiggles (GNW) are graphene-based nanostructures obtained by making alternated regular cuts in pristine graphene nanoribbons. GNW were recently synthesized and it was demonstrated that they exhibit tunable electronic and magnetic properties by just varying their shape. Here, we have investigated the mechanical properties and fracture patterns of a large number of GNW of different shapes and sizes using fully atomistic reactive molecular dynamics simulations. Our results show that the GNW mechanical properties are strongly dependent on its shape and size and, as a general trend narrow sheets have larger ultimate strength and Young's modulus than wide ones. The estimated Young's modulus values were found to be in a range of ≈100−1000 GPa and the ultimate strength in a range of ≈20−110 GPa, depending on GNW shape. Also, super-ductile behavior under strain was observed for some structures.},
note = {See also ArxIv version: https://arxiv.org/abs/1702.01100},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Bizao, RA; Botari, T; Galvao, DS
Mechanical Properties of Graphene Nanowiggles Proceedings
Cambridge University Press, vol. 1658, 2014.
@proceedings{bizao2014mechanical,
title = {Mechanical Properties of Graphene Nanowiggles},
author = {Bizao, RA and Botari, T and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=9248042&fileId=S1946427414004023},
year = {2014},
date = {2014-01-01},
journal = {MRS Proceedings},
volume = {1658},
pages = {mrsf13--1658},
publisher = {Cambridge University Press},
abstract = {In this work we have investigated the mechanical properties and fracture patterns of some graphene nanowiggles (GNWs). Graphene nanoribbons are finite graphene segments with a large aspect ratio, while GNWs are nonaligned periodic repetitions of graphene nanoribbons. We have carried out fully atomistic molecular dynamics simulations using a reactive force field (ReaxFF), as implemented in the LAMPPS (Large-scale Atomic/Molecular Massively Parallel Simulator) code. Our results showed that the GNW fracture patterns are strongly dependent on the nanoribbon topology and present an interesting behavior, since some narrow sheets have larger ultimate failure strain values. This can be explained by the fact that narrow nanoribbons have more angular freedom when compared to wider ones, which can create a more efficient way to accumulate and to dissipate strain/stress. We have also observed the formation of linear atomic chains (LACs) and some structural defect reconstructions during the material rupture. The reported graphene failure patterns, where zigzag/armchair edge terminated graphene structures are fractured along armchair/zigzag lines, were not observed in the GNW analyzed cases.},
keywords = {},
pubstate = {published},
tppubtype = {proceedings}
}
Martins, BVC; Galvao, DS
Curved graphene nanoribbons: structure and dynamics of carbon nanobelts Journal Article
In: Nanotechnology, vol. 21, no. 7, pp. 075710, 2010.
@article{martins2010curved,
title = {Curved graphene nanoribbons: structure and dynamics of carbon nanobelts},
author = {Martins, BVC and Galvao, DS},
url = {http://iopscience.iop.org/0957-4484/21/7/075710},
year = {2010},
date = {2010-01-01},
journal = {Nanotechnology},
volume = {21},
number = {7},
pages = {075710},
publisher = {IOP Publishing},
abstract = {Carbon nanoribbons (CNRs) are graphene (planar) structures with a large aspect ratio. Carbon nanobelts (CNBs) are small graphene nanoribbons rolled up into spiral-like structures, i.e. carbon nanoscrolls (CNSs) with a large aspect ratio. In this work we investigated the energetics and dynamical aspects of CNBs formed from rolling up CNRs. We have carried out molecular dynamics simulations using reactive empirical bond-order potentials. Our results show that, similarly to CNSs, CNB formation is dominated by two major energy contributions, the increase in the elastic energy due to the bending of the initial planar configuration (decreasing structural stability) and the energetic gain due to van der Waals interactions of the overlapping surface of the rolled layers (increasing structural stability). Beyond a critical diameter value these scrolled structures can be even more stable (in terms of energy) than their equivalent planar configurations. In contrast to CNSs that require energy-assisted processes (sonication, chemical reactions, etc) to be formed, CNBs can be spontaneously formed from low temperature driven processes. Long CNBs (length of ~30.0 nm) tend to exhibit self-folded racket-like conformations with formation dynamics very similar to the one observed for long carbon nanotubes. Shorter CNBs will be more likely to form perfect scrolled structures. Possible synthetic routes to fabricate CNBs from graphene membranes are also addressed.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Caetano, Ewerton WS; Freire, Valder N; dos Santos, Sergio G; Albuquerque, EL; Galvao, Douglas S; Sato, Fernando
Defects in Graphene-Based Twisted Nanoribbons: Structural, Electronic, and Optical Properties Journal Article
In: Langmuir, vol. 25, no. 8, pp. 4751–4759, 2009.
@article{caetano2009defects,
title = {Defects in Graphene-Based Twisted Nanoribbons: Structural, Electronic, and Optical Properties},
author = {Caetano, Ewerton WS and Freire, Valder N and dos Santos, Sergio G and Albuquerque, EL and Galvao, Douglas S and Sato, Fernando},
url = {http://pubs.acs.org/doi/abs/10.1021/la803929f},
year = {2009},
date = {2009-01-01},
journal = {Langmuir},
volume = {25},
number = {8},
pages = {4751--4759},
publisher = {ACS Publications},
abstract = {We present some computational simulations of graphene-based nanoribbons with a number of half-twists varying from 0 to 4 and two types of defects obtained by removing a single carbon atom from two different sites. Optimized geometries are found by using a mix of classical quantum semiempirical computations. According with the simulations results, the local curvature of the nanoribbons increases at the defect sites, especially for a higher number of half-twists. The HOMO−LUMO energy gap of the nanostructures has significant variation when the number of half-twists increases for the defective nanoribbons. At the quantum semiempirical level, the first optically active transitions and oscillator strengths are calculated using the full configuration interaction (CI) framework, and the optical absorption in the UV/vis range (electronic transitions) and in the infrared (vibrational transitions) are achieved. Distinct nanoribbons show unique spectral signatures in the UV/vis range, with the first absorption peaks in wavelengths ranging from the orange to the violet. Strong absorption is observed in the ultraviolet region, although differences in their infrared spectra are hardly discernible.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Caetano, Ewerton WS; Freire, Valder N; Santos, Sergio G dos; Galvao, Douglas S; Sato, Fernando
Mobius and twisted graphene nanoribbons: stability, geometry and electronic properties Journal Article
In: arXiv preprint arXiv:0903.2080, 2009.
@article{caetano2009m,
title = {Mobius and twisted graphene nanoribbons: stability, geometry and electronic properties},
author = {Caetano, Ewerton WS and Freire, Valder N and Santos, Sergio G dos and Galvao, Douglas S and Sato, Fernando},
url = {http://arxiv.org/abs/0903.2080},
year = {2009},
date = {2009-01-01},
journal = {arXiv preprint arXiv:0903.2080},
abstract = {Results of classical force field geometry optimizations for twisted graphene nanoribbons with a number of twists Nt varying from 0 to 7 (the case Nt=1 corresponds to a half-twist M"obius nanoribbon) are presented in this work. Their structural stability was investigated using the Brenner reactive force field. The best classical molecular geometries were used as input for semiempirical calculations, from which the electronic properties (energy levels, HOMO, LUMO orbitals) were computed for each structure. CI wavefunctions were also calculated in the complete active space framework taking into account eigenstates from HOMO-4 to LUMO+4, as well as the oscillator strengths corresponding to the first optical transitions in the UV-VIS range. The lowest energy molecules were found less symmetric than initial configurations, and the HOMO-LUMO energy gaps are larger than the value found for the nanographene used to build them due to electronic localization effects created by the twisting. A high number of twists leads to a sharp increase of the HOMO → LUMO transition energy. We suggest that some twisted nanoribbons could form crystals stabilized by dipolar interactions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
E. W. S.; Freire Caetano, V. N. ; dos Santos
Mobius and twisted graphene nanoribbons: Stability, geometry, and electronic properties Journal Article
In: THE JOURNAL OF CHEMICAL PHYSICS, vol. 128, pp. 164719, 2008.
@article{Caetano2008,
title = {Mobius and twisted graphene nanoribbons: Stability, geometry, and electronic properties},
author = {Caetano, E. W. S.; Freire, V. N.; dos Santos, S. G.; Galvao, D. S.,and Sato, F.},
url = {http://scitation.aip.org/content/aip/journal/jcp/128/16/10.1063/1.2908739},
year = {2008},
date = {2008-04-29},
journal = {THE JOURNAL OF CHEMICAL PHYSICS},
volume = {128},
pages = {164719},
abstract = {Results of classical force field geometry optimizations for twisted graphenenanoribbons with a number of twists Nt varying from 0 to 7 (the case Nt=1 corresponds to a half-twist Möbius nanoribbon) are presented in this work. Their structural stability was investigated using the Brenner reactive force field. The best classical molecular geometries were used as input for semiempirical calculations, from which the electronic properties (energy levels, HOMO, LUMO orbitals) were computed for each structure. CI wavefunctions were also calculated in the complete active space framework taking into account eigenstates from HOMO−4 to LUMO+4, as well as the oscillator strengths corresponding to the first optical transitions in the UV-VIS range. The lowest energy molecules were found less symmetric than initial configurations, and the HOMO-LUMO energy gaps are larger than the value found for the nanographene used to build them due to electronic localization effects created by the twisting. A high number of twists leads to a sharp increase of the HOMO→LUMO transition energy. We suggest that some twisted nanoribbons could form crystals stabilized by dipolar interactions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2017
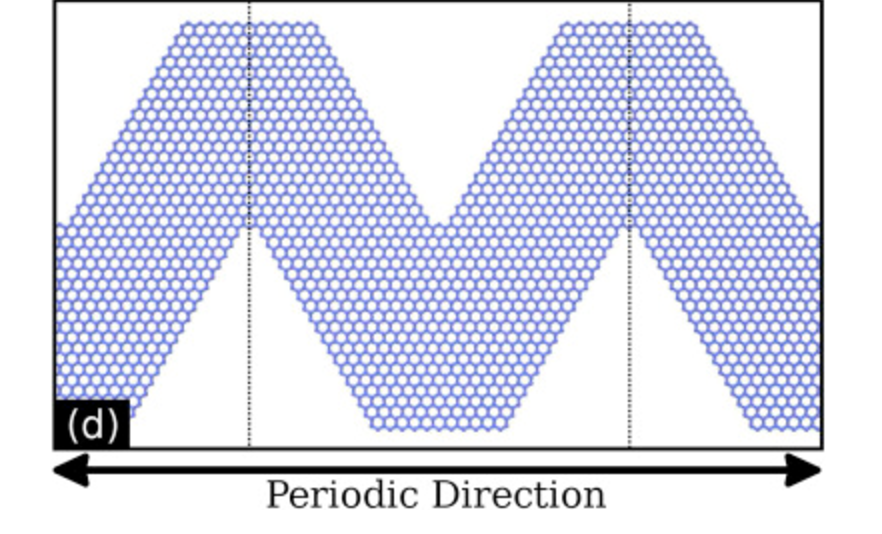
Bizao, Rafael A; Botari, Tiago; Perim, Eric; Pugno, Nicola M; Galvao, Douglas S
Mechanical properties and fracture patterns of graphene (graphitic) nanowiggles Journal Article
In: Carbon, vol. 119, pp. 431-437, 2017, (See also ArxIv version: https://arxiv.org/abs/1702.01100).
Abstract | Links | BibTeX | Tags: Graphene, Molecular Dynamics, NanoRibbons, Nanowiggles
@article{Bizao2017b,
title = {Mechanical properties and fracture patterns of graphene (graphitic) nanowiggles},
author = {Bizao, Rafael A and Botari, Tiago and Perim, Eric and Pugno, Nicola M and Galvao, Douglas S},
url = {http://www.sciencedirect.com/science/article/pii/S0008622317303743},
doi = {10.1016/j.carbon.2017.04.018},
year = {2017},
date = {2017-04-14},
journal = {Carbon},
volume = {119},
pages = {431-437},
abstract = {Graphene nanowiggles (GNW) are graphene-based nanostructures obtained by making alternated regular cuts in pristine graphene nanoribbons. GNW were recently synthesized and it was demonstrated that they exhibit tunable electronic and magnetic properties by just varying their shape. Here, we have investigated the mechanical properties and fracture patterns of a large number of GNW of different shapes and sizes using fully atomistic reactive molecular dynamics simulations. Our results show that the GNW mechanical properties are strongly dependent on its shape and size and, as a general trend narrow sheets have larger ultimate strength and Young's modulus than wide ones. The estimated Young's modulus values were found to be in a range of ≈100−1000 GPa and the ultimate strength in a range of ≈20−110 GPa, depending on GNW shape. Also, super-ductile behavior under strain was observed for some structures.},
note = {See also ArxIv version: https://arxiv.org/abs/1702.01100},
keywords = {Graphene, Molecular Dynamics, NanoRibbons, Nanowiggles},
pubstate = {published},
tppubtype = {article}
}
2014
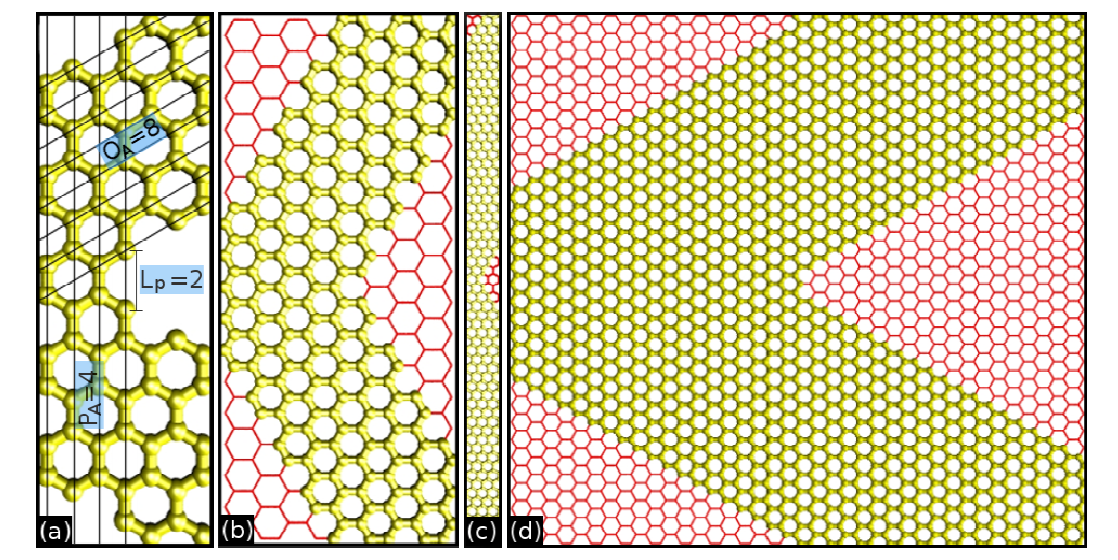
Bizao, RA; Botari, T; Galvao, DS
Mechanical Properties of Graphene Nanowiggles Proceedings
Cambridge University Press, vol. 1658, 2014.
Abstract | Links | BibTeX | Tags: Graphene, Molecular Dynamics, NanoRibbons, Nanowiggles
@proceedings{bizao2014mechanical,
title = {Mechanical Properties of Graphene Nanowiggles},
author = {Bizao, RA and Botari, T and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=9248042&fileId=S1946427414004023},
year = {2014},
date = {2014-01-01},
journal = {MRS Proceedings},
volume = {1658},
pages = {mrsf13--1658},
publisher = {Cambridge University Press},
abstract = {In this work we have investigated the mechanical properties and fracture patterns of some graphene nanowiggles (GNWs). Graphene nanoribbons are finite graphene segments with a large aspect ratio, while GNWs are nonaligned periodic repetitions of graphene nanoribbons. We have carried out fully atomistic molecular dynamics simulations using a reactive force field (ReaxFF), as implemented in the LAMPPS (Large-scale Atomic/Molecular Massively Parallel Simulator) code. Our results showed that the GNW fracture patterns are strongly dependent on the nanoribbon topology and present an interesting behavior, since some narrow sheets have larger ultimate failure strain values. This can be explained by the fact that narrow nanoribbons have more angular freedom when compared to wider ones, which can create a more efficient way to accumulate and to dissipate strain/stress. We have also observed the formation of linear atomic chains (LACs) and some structural defect reconstructions during the material rupture. The reported graphene failure patterns, where zigzag/armchair edge terminated graphene structures are fractured along armchair/zigzag lines, were not observed in the GNW analyzed cases.},
keywords = {Graphene, Molecular Dynamics, NanoRibbons, Nanowiggles},
pubstate = {published},
tppubtype = {proceedings}
}
2010
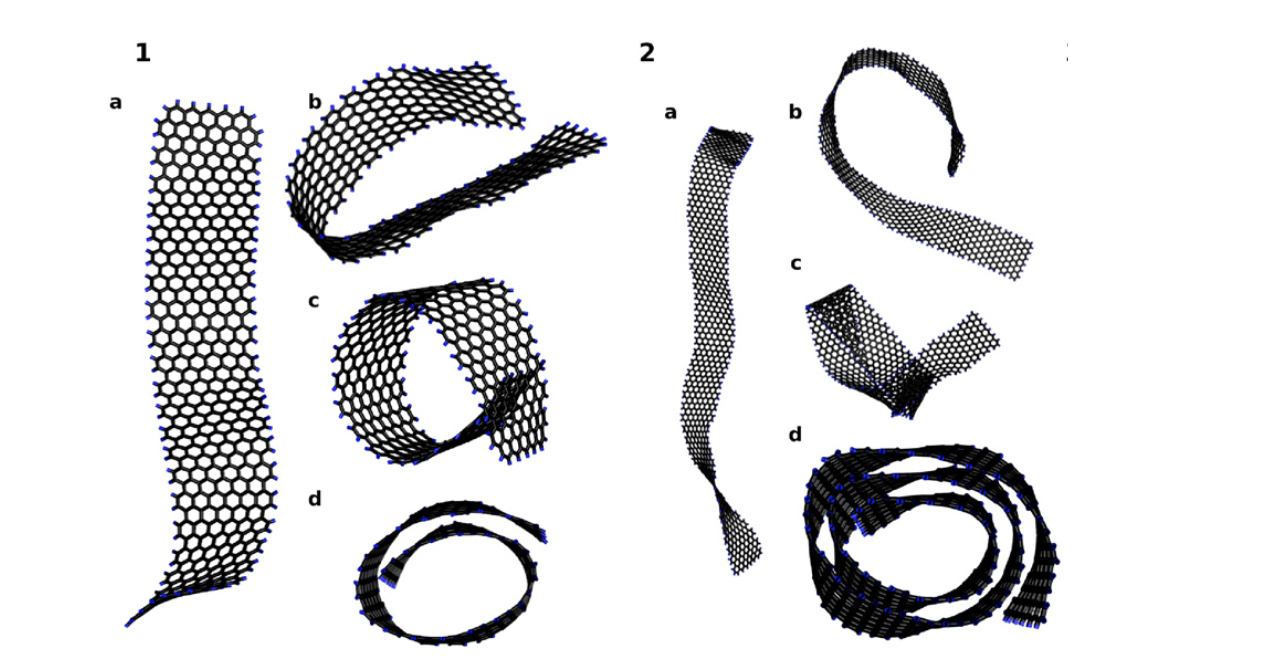
Martins, BVC; Galvao, DS
Curved graphene nanoribbons: structure and dynamics of carbon nanobelts Journal Article
In: Nanotechnology, vol. 21, no. 7, pp. 075710, 2010.
Abstract | Links | BibTeX | Tags: Folding, Graphene, Mechanical Properties, Nanobelts, NanoRibbons
@article{martins2010curved,
title = {Curved graphene nanoribbons: structure and dynamics of carbon nanobelts},
author = {Martins, BVC and Galvao, DS},
url = {http://iopscience.iop.org/0957-4484/21/7/075710},
year = {2010},
date = {2010-01-01},
journal = {Nanotechnology},
volume = {21},
number = {7},
pages = {075710},
publisher = {IOP Publishing},
abstract = {Carbon nanoribbons (CNRs) are graphene (planar) structures with a large aspect ratio. Carbon nanobelts (CNBs) are small graphene nanoribbons rolled up into spiral-like structures, i.e. carbon nanoscrolls (CNSs) with a large aspect ratio. In this work we investigated the energetics and dynamical aspects of CNBs formed from rolling up CNRs. We have carried out molecular dynamics simulations using reactive empirical bond-order potentials. Our results show that, similarly to CNSs, CNB formation is dominated by two major energy contributions, the increase in the elastic energy due to the bending of the initial planar configuration (decreasing structural stability) and the energetic gain due to van der Waals interactions of the overlapping surface of the rolled layers (increasing structural stability). Beyond a critical diameter value these scrolled structures can be even more stable (in terms of energy) than their equivalent planar configurations. In contrast to CNSs that require energy-assisted processes (sonication, chemical reactions, etc) to be formed, CNBs can be spontaneously formed from low temperature driven processes. Long CNBs (length of ~30.0 nm) tend to exhibit self-folded racket-like conformations with formation dynamics very similar to the one observed for long carbon nanotubes. Shorter CNBs will be more likely to form perfect scrolled structures. Possible synthetic routes to fabricate CNBs from graphene membranes are also addressed.
},
keywords = {Folding, Graphene, Mechanical Properties, Nanobelts, NanoRibbons},
pubstate = {published},
tppubtype = {article}
}
2009
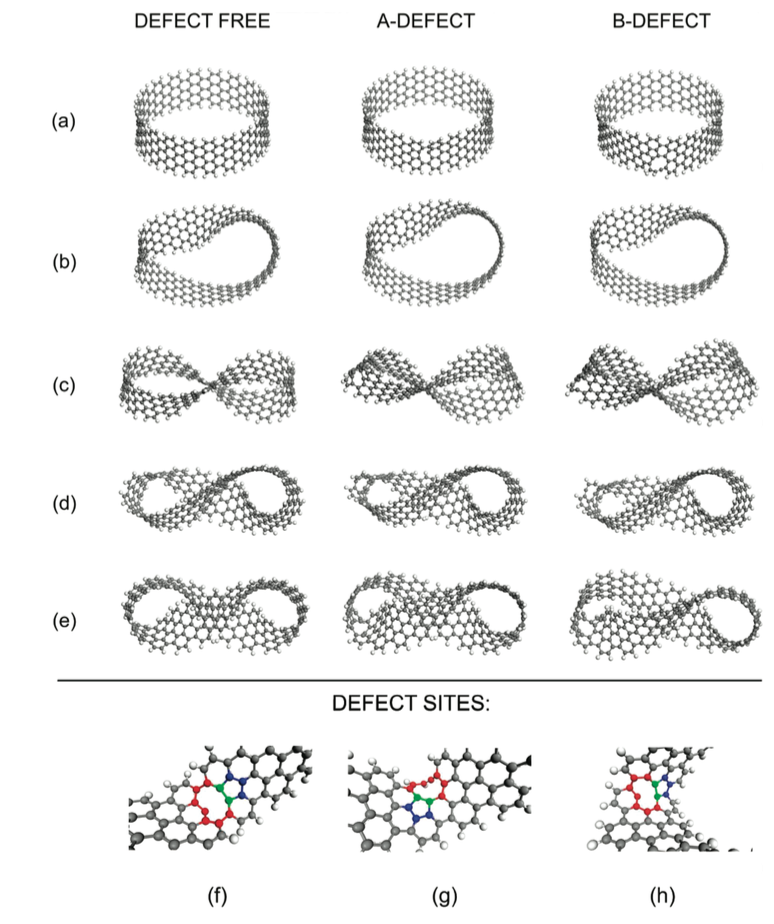
Caetano, Ewerton WS; Freire, Valder N; dos Santos, Sergio G; Albuquerque, EL; Galvao, Douglas S; Sato, Fernando
Defects in Graphene-Based Twisted Nanoribbons: Structural, Electronic, and Optical Properties Journal Article
In: Langmuir, vol. 25, no. 8, pp. 4751–4759, 2009.
Abstract | Links | BibTeX | Tags: Defects, Mobius, NanoRibbons, Twisting
@article{caetano2009defects,
title = {Defects in Graphene-Based Twisted Nanoribbons: Structural, Electronic, and Optical Properties},
author = {Caetano, Ewerton WS and Freire, Valder N and dos Santos, Sergio G and Albuquerque, EL and Galvao, Douglas S and Sato, Fernando},
url = {http://pubs.acs.org/doi/abs/10.1021/la803929f},
year = {2009},
date = {2009-01-01},
journal = {Langmuir},
volume = {25},
number = {8},
pages = {4751--4759},
publisher = {ACS Publications},
abstract = {We present some computational simulations of graphene-based nanoribbons with a number of half-twists varying from 0 to 4 and two types of defects obtained by removing a single carbon atom from two different sites. Optimized geometries are found by using a mix of classical quantum semiempirical computations. According with the simulations results, the local curvature of the nanoribbons increases at the defect sites, especially for a higher number of half-twists. The HOMO−LUMO energy gap of the nanostructures has significant variation when the number of half-twists increases for the defective nanoribbons. At the quantum semiempirical level, the first optically active transitions and oscillator strengths are calculated using the full configuration interaction (CI) framework, and the optical absorption in the UV/vis range (electronic transitions) and in the infrared (vibrational transitions) are achieved. Distinct nanoribbons show unique spectral signatures in the UV/vis range, with the first absorption peaks in wavelengths ranging from the orange to the violet. Strong absorption is observed in the ultraviolet region, although differences in their infrared spectra are hardly discernible.},
keywords = {Defects, Mobius, NanoRibbons, Twisting},
pubstate = {published},
tppubtype = {article}
}
Caetano, Ewerton WS; Freire, Valder N; Santos, Sergio G dos; Galvao, Douglas S; Sato, Fernando
Mobius and twisted graphene nanoribbons: stability, geometry and electronic properties Journal Article
In: arXiv preprint arXiv:0903.2080, 2009.
Abstract | Links | BibTeX | Tags: Graphene, Mobius, NanoRibbons, Structure
@article{caetano2009m,
title = {Mobius and twisted graphene nanoribbons: stability, geometry and electronic properties},
author = {Caetano, Ewerton WS and Freire, Valder N and Santos, Sergio G dos and Galvao, Douglas S and Sato, Fernando},
url = {http://arxiv.org/abs/0903.2080},
year = {2009},
date = {2009-01-01},
journal = {arXiv preprint arXiv:0903.2080},
abstract = {Results of classical force field geometry optimizations for twisted graphene nanoribbons with a number of twists Nt varying from 0 to 7 (the case Nt=1 corresponds to a half-twist M"obius nanoribbon) are presented in this work. Their structural stability was investigated using the Brenner reactive force field. The best classical molecular geometries were used as input for semiempirical calculations, from which the electronic properties (energy levels, HOMO, LUMO orbitals) were computed for each structure. CI wavefunctions were also calculated in the complete active space framework taking into account eigenstates from HOMO-4 to LUMO+4, as well as the oscillator strengths corresponding to the first optical transitions in the UV-VIS range. The lowest energy molecules were found less symmetric than initial configurations, and the HOMO-LUMO energy gaps are larger than the value found for the nanographene used to build them due to electronic localization effects created by the twisting. A high number of twists leads to a sharp increase of the HOMO → LUMO transition energy. We suggest that some twisted nanoribbons could form crystals stabilized by dipolar interactions.},
keywords = {Graphene, Mobius, NanoRibbons, Structure},
pubstate = {published},
tppubtype = {article}
}
2008

E. W. S.; Freire Caetano, V. N. ; dos Santos
Mobius and twisted graphene nanoribbons: Stability, geometry, and electronic properties Journal Article
In: THE JOURNAL OF CHEMICAL PHYSICS, vol. 128, pp. 164719, 2008.
Abstract | Links | BibTeX | Tags: DFT, Graphene, Mobis, NanoRibbons, Structure
@article{Caetano2008,
title = {Mobius and twisted graphene nanoribbons: Stability, geometry, and electronic properties},
author = {Caetano, E. W. S.; Freire, V. N.; dos Santos, S. G.; Galvao, D. S.,and Sato, F.},
url = {http://scitation.aip.org/content/aip/journal/jcp/128/16/10.1063/1.2908739},
year = {2008},
date = {2008-04-29},
journal = {THE JOURNAL OF CHEMICAL PHYSICS},
volume = {128},
pages = {164719},
abstract = {Results of classical force field geometry optimizations for twisted graphenenanoribbons with a number of twists Nt varying from 0 to 7 (the case Nt=1 corresponds to a half-twist Möbius nanoribbon) are presented in this work. Their structural stability was investigated using the Brenner reactive force field. The best classical molecular geometries were used as input for semiempirical calculations, from which the electronic properties (energy levels, HOMO, LUMO orbitals) were computed for each structure. CI wavefunctions were also calculated in the complete active space framework taking into account eigenstates from HOMO−4 to LUMO+4, as well as the oscillator strengths corresponding to the first optical transitions in the UV-VIS range. The lowest energy molecules were found less symmetric than initial configurations, and the HOMO-LUMO energy gaps are larger than the value found for the nanographene used to build them due to electronic localization effects created by the twisting. A high number of twists leads to a sharp increase of the HOMO→LUMO transition energy. We suggest that some twisted nanoribbons could form crystals stabilized by dipolar interactions.},
keywords = {DFT, Graphene, Mobis, NanoRibbons, Structure},
pubstate = {published},
tppubtype = {article}
}
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ

