
Malviya, Kirtman D; Oliveira, Eliezer F; Autreto, Pedro A S; Ajayan, Pulickel M; Galvao, D S; Tiwary, Candra S; Chattopadhyay, Kumanio
Mixing the immiscible through high-velocity mechanical impacts: an experimental and theoretical study Journal Article
In: Journal of Physics D: Applied Physics, vol. 52, no. 44, pp. 445304, 2019.
@article{Malviya2019,
title = {Mixing the immiscible through high-velocity mechanical impacts: an experimental and theoretical study},
author = {Malviya, Kirtman D and Oliveira, Eliezer F and Autreto, Pedro A S and Ajayan, Pulickel M and Galvao, D S and Tiwary, Candra S and Chattopadhyay, Kumanio},
url = {https://iopscience.iop.org/article/10.1088/1361-6463/ab36d1/meta},
doi = {10.1088/1361-6463/ab36d1},
year = {2019},
date = {2019-08-20},
journal = {Journal of Physics D: Applied Physics},
volume = {52},
number = {44},
pages = {445304},
abstract = {In two-component metallic systems, thermodynamic immiscibility leads to phase separation
such as in two-phase eutectic compositional alloys. The limit of the immiscibility of
component elements under non-equilibrium conditions have been explored, but achieving
complete miscibility and formation of single phase microstructures in eutectic alloys would
be unprecedented. Here we report that during low-temperature ball milling that provides high
energy impact, complete mixing of phases can occur in immiscible Ag-Cu eutectic alloys.
From combined theoretical and experimental studies, we show that impact can produce solid
solutions of Ag-Cu nanoparticles of eutectic composition. Our results show that phase
diagrams of low dimensional materials under non-equilibrium conditions remain unexplored
and could lead to new alloy microstructures drastically different from their bulk counterparts.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
such as in two-phase eutectic compositional alloys. The limit of the immiscibility of
component elements under non-equilibrium conditions have been explored, but achieving
complete miscibility and formation of single phase microstructures in eutectic alloys would
be unprecedented. Here we report that during low-temperature ball milling that provides high
energy impact, complete mixing of phases can occur in immiscible Ag-Cu eutectic alloys.
From combined theoretical and experimental studies, we show that impact can produce solid
solutions of Ag-Cu nanoparticles of eutectic composition. Our results show that phase
diagrams of low dimensional materials under non-equilibrium conditions remain unexplored
and could lead to new alloy microstructures drastically different from their bulk counterparts.
de Sousa, Jose Moreira; Autreto, Pedro da Silva; Galvao, Douglas Soares
Hydrogenation Dynamics Process of Single-wall Carbon Nanotube Twisted (under review) Journal Article
In: 2019.
@article{deSousa2019d,
title = {Hydrogenation Dynamics Process of Single-wall Carbon Nanotube Twisted (under review)},
author = {de Sousa, Jose Moreira and Autreto, Pedro da Silva and Galvao, Douglas Soares},
year = {2019},
date = {2019-07-15},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Eliezer F; Autreto Oliveira, Pedro AS; Woellner
On the mechanical properties of protomene: A theoretical investigation Journal Article
In: Computational Materials Science, vol. 161, pp. 190-198, 2019.
@article{Oliveira2019c,
title = {On the mechanical properties of protomene: A theoretical investigation},
author = {Oliveira, Eliezer F; Autreto, Pedro AS; Woellner, Cristiano F; Galvao, Douglas S},
year = {2019},
date = {2019-02-07},
journal = {Computational Materials Science},
volume = {161},
pages = {190-198},
abstract = {We report a detailed study through fully atomistic molecular dynamics simulations and DFT calculations on the mechanical properties of protomene. Protomene is a new carbon allotrope composed of a mixture of sp2 and sp3 hybridized states. Our results indicate that protomene presents an anisotropic behavior about tensile deformations. At room temperature, protomene presents an ultimate strength of ~100 GPa and Young's modulus of ~600 GPa, lower than the same for other carbon allotropes. Despite that, protomente presents the highest ultimate strain along the z-direction (~ 24.7%). Our results also show that stretching the protomene along the z-direction or heating it can induce a semiconductor-metallic phase transition, due to a high amount of sp3 bonds that are converted to sp2 ones.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Sanjit; Ozden Bhowmick, Sehmus; Bizão
High temperature quasistatic and dynamic mechanical behavior of interconnected 3D carbon nanotube structures Journal Article
In: Carbon, vol. 142, pp. 291-299, 2019.
@article{Bhowmick2019,
title = {High temperature quasistatic and dynamic mechanical behavior of interconnected 3D carbon nanotube structures},
author = {Bhowmick, Sanjit; Ozden, Sehmus; Bizão, Rafael A; Machado, Leonardo Dantas; Asif, SA Syed; Pugno, Nicola M; Galvao, Douglas S; Tiwary, Chandra Sekhar; Ajayan, PM},
url = {https://www.sciencedirect.com/science/article/pii/S0008622318308911},
doi = {10.1016/j.carbon.2018.09.075},
year = {2019},
date = {2019-02-01},
journal = {Carbon},
volume = {142},
pages = {291-299},
abstract = {Carbon nanotubes (CNTs) are one of the most appealing materials in recent history for both research and commercial interest because of their outstanding physical, chemical, and electrical properties. This is particularly true for 3D arrangements of CNTs which enable their use in larger scale devices and structures. In this paper, the effect of temperature on the quasistatic and dynamic deformation behavior of 3D CNT structures is presented for the first time. An in situ high-temperature nanomechanical instrument was used inside an SEM at high vacuum to investigate mechanical properties of covalently interconnected CNT porous structures in a wide range of temperature. An irreversible bucking at the base of pillar samples was found as a major mode of deformation at room and elevated temperatures. It has been observed that elastic modulus and critical load to first buckle formation decrease progressively with increasing temperature from 25 °C to 750 °C. To understand fatigue resistance, pillars made from this unique structure were compressed to 100 cycles at room temperature and 750 °C. While the structure showed remarkable resistance to fatigue at room temperature, high temperature significantly lowers fatigue resistance. Molecular dynamics (MD) simulation of compression highlights the critical role played by covalent interconnections which prevent localized bending and improve mechanical properties.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Fonseca, Alexandre F.; Galvao, Douglas S.
Self-tearing and self-peeling of folded graphene nanoribbons Journal Article
In: Carbon, vol. 143, pp. 230-239, 2019.
@article{Fonseca2019,
title = {Self-tearing and self-peeling of folded graphene nanoribbons},
author = {Alexandre F. Fonseca and Douglas S. Galvao},
url = {https://www.sciencedirect.com/science/article/pii/S0008622318310431},
doi = {10.1016/j.carbon.2018.11.020},
year = {2019},
date = {2019-01-05},
journal = {Carbon},
volume = {143},
pages = {230-239},
abstract = {A recent experimental study showed that an induced folded flap of graphene can spontaneously drive itself its tearing and peeling off a substrate, thus producing long, micrometer sized, regular trapezoidal-shaped folded graphene nanoribbons. As long as the size of the graphene flaps is above a threshold value, the “tug of war” between the forces of adhesion of graphene-graphene and graphene-substrate, flexural strain of folded region and carbon-carbon (C-C) covalent bonds favor the self-tearing and self-peeling off process. As the detailed information regarding the atomic scale mechanism involved in the process remains not fully understood, we carried out atomistic reactive molecular dynamics simulations to address some features of the process. We show that large thermal fluctuations can prevent the process by increasing the probability of chemical reactions between carbon dangling bonds of adjacent graphene layers. The effects of the strength of attraction between graphene and the substrate on the ribbon growth velocities at the early stages of the phenomenon were also investigated. Structures with initial armchair crack-edges were observed to form more uniform cuts than those having initial zigzag ones. Our results are of importance to help set up new experiments on this phenomenon, especially with samples with nanoscale sized cuts. },
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Eliezer F; Autreto Oliveira, Pedro AS; Woellner
Mechanical Properties of Protomene: A Molecular Dynamics Investigation Journal Article
In: MRS Advances, 2019.
@article{Oliveira2019,
title = {Mechanical Properties of Protomene: A Molecular Dynamics Investigation},
author = {Oliveira, Eliezer F; Autreto, Pedro AS; Woellner, Cristiano F; Galvao, Douglas S},
url = {www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-protomene-a-molecular-dynamics-investigation/CBAC89BDB5942E3353A5C00BD5D0D9CA},
doi = {10.1557/adv.2018.670},
year = {2019},
date = {2019-01-05},
journal = {MRS Advances},
abstract = {Recently, a new class of carbon allotrope called protomene was proposed. This new structure is composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3 carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now, its mechanical properties have not been investigated. In this work, we have investigated protomene mechanical behavior under tensile strain through fully atomistic reactive molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS code. At room temperature, our results show that the protomene is very stable and the obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical fracture.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Pedro AS Autreto Eliezer F Oliveira, Cristiano F Woellner
Mechanical Properties of Protomene: A Molecular Dynamics Investigation Online
2018, (preprint arXiv:1810.09924v1 ).
@online{Oliveira2018g,
title = {Mechanical Properties of Protomene: A Molecular Dynamics Investigation},
author = {Eliezer F Oliveira, Pedro AS Autreto, Cristiano F Woellner, Douglas S Galvao},
url = {https://arxiv.org/abs/1810.09924},
year = {2018},
date = {2018-10-23},
abstract = {Recently, a new class of carbon allotrope called protomene was proposed. This new structure is
composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3
carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations
have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now,
its mechanical properties have not been investigated. In this work, we have investigated
protomene mechanical behavior under tensile strain through fully atomistic reactive
molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS
code. At room temperature, our results show that the protomene is very stable and the
obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest
ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate
strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical
fracture.},
note = {preprint arXiv:1810.09924v1 },
keywords = {},
pubstate = {published},
tppubtype = {online}
}
composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3
carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations
have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now,
its mechanical properties have not been investigated. In this work, we have investigated
protomene mechanical behavior under tensile strain through fully atomistic reactive
molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS
code. At room temperature, our results show that the protomene is very stable and the
obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest
ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate
strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical
fracture.
Alexandre F. Fonseca, Douglas S. Galvao
Self-tearing and self-peeling of folded graphene nanoribbons Online
2018, (preprint arXiv:1808.08872).
@online{Fonseca2018d,
title = { Self-tearing and self-peeling of folded graphene nanoribbons},
author = {Alexandre F. Fonseca, Douglas S. Galvao
},
url = {https://arxiv.org/abs/1808.08872},
year = {2018},
date = {2018-08-27},
abstract = {A recent experimental study showed that an induced folded flap of graphene can spontaneously drive itself its tearing and peeling off a substrate, thus producing long, micrometer sized, regular trapezoidal-shaped folded graphene nanoribbons. As long as the size of the graphene flaps is above a threshold value, the 'tug of war' between the forces of adhesion of graphene-graphene and graphene-substrate, flexural strain of folded region and carbon-carbon (C-C) covalent bonds favor the self-tearing and self-peeling off process. As the detailed information regarding the atomic scale mechanism involved in the process remains not fully understood, we carried out atomistic reactive molecular dynamics simulations to address some features of the process. We show that large thermal fluctuations can prevent the process by increasing the probability of chemical reactions between carbon dangling bonds of adjacent graphene layers. The effects of the strength of attraction between graphene and the substrate on the ribbon growth velocities at the early stages of the phenomenon were also investigated. Structures with initial armchair crack-edges were observed to form more uniform cuts than those having initial zigzag ones. Our results are of importance to help set up new experiments on this phenomenon, especially with samples with nanoscale sized cuts.},
note = {preprint arXiv:1808.08872},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Borges, Daiane Damasceno; Galvao, Douglas S.
Schwarzites for Natural Gas Storage: A Grand-Canonical Monte Carlo Study Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 115-120, 2018.
@article{Borges2018d,
title = {Schwarzites for Natural Gas Storage: A Grand-Canonical Monte Carlo Study },
author = {Daiane Damasceno Borges and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/schwarzites-for-natural-gas-storage-a-grandcanonical-monte-carlo-study/2DF8D601AF8EF04BBAC5CCCBEFA8339E},
doi = {https://doi.org/10.1557/adv.2018.190},
year = {2018},
date = {2018-02-13},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = {115-120},
abstract = {he 3D porous carbon-based structures called Schwarzites have been recently a subject of renewed interest due to the possibility of being synthesized in the near future. These structures exhibit negatively curvature topologies with tuneable porous sizes and shapes, which make them natural candidates for applications such as CO2 capture, gas storage and separation. Nevertheless, the adsorption properties of these materials have not been fully investigated. Following this motivation, we have carried out Grand-Canonical Monte Carlo simulations to study the adsorption of small molecules such as CO2, CO, CH4, N2 and H2, in a series of Schwarzites structures. Here, we present our preliminary results on natural gas adsorptive capacity in association with analyses of the guest-host interaction strengths. Our results show that Schwarzites P7par, P8bal and IWPg are the most promising structures with very high CO2 and CH4 adsorption capacity and low saturation pressure (<1bar) at ambient temperature. The P688 is interesting for H2 storage due to its exceptional high H2 adsorption enthalpy value of -19kJ/mol.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Woellner, Cristiano F.; Botari, Tiago; Perim, Eric; Galvao, Douglas S.
Mechanical Properties of Schwarzites - A Fully Atomistic Reactive Molecular Dynamics Investigation Journal Article
In: MRS Advances, pp. 1-6, 2018.
@article{Woellner2018b,
title = {Mechanical Properties of Schwarzites - A Fully Atomistic Reactive Molecular Dynamics Investigation},
author = {Cristiano F. Woellner and Tiago Botari and Eric Perim and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-schwarzites-a-fully-atomistic-reactive-molecular-dynamics-investigation/012AF477491A46541A052C944E4E4834},
doi = { https://doi.org/10.1557/adv.2018.124},
year = {2018},
date = {2018-01-29},
journal = {MRS Advances},
pages = {1-6},
abstract = {Schwarzites are crystalline, 3D porous structures with a stable negative curvature formed of sp2-hybridized carbon atoms. These structures present topologies with tunable porous size and shape and unusual mechanical properties. In this work, we have investigated the mechanical behavior under compressive strain and energy absorption of four different Schwarzites. We considered two Schwarzites families, the so-called Gyroid and Primitive and two structures from each family. We carried out reactive molecular dynamics simulations, using the ReaxFF force field as available in the LAMMPS code. Our results also show they exhibit remarkable resilience under mechanical compression. They can be reduced to half of their original size before structural failure (fracture) occurs.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Woellner, Cristiano F.; Owuor, Peter S.; Li, Tong; Vinod, Soumya; Ozden, Sehmus; Kosolwattana, Suppanat; Bhowmick, Sanjit; Duy, Luong X.; Salvatierra, Rodrigo V.; Wei, Bingqing; Asif, Syed A. S.; Tour, James M.; Vajtai, Robert; Lou, Jun; Galvão, Douglas S.; Tiwary, Chandra S.; Ajayan, Pulickel. M.
Mechanical Properties of Ultralow Density Graphene Oxide/Polydimethylsiloxane Foams Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 61-66, 2018.
@article{Woellner2018c,
title = {Mechanical Properties of Ultralow Density Graphene Oxide/Polydimethylsiloxane Foams},
author = {Cristiano F. Woellner and Peter S. Owuor and Tong Li and Soumya Vinod and Sehmus Ozden and Suppanat Kosolwattana and Sanjit Bhowmick and Luong X. Duy and Rodrigo V. Salvatierra and Bingqing Wei and Syed A. S. Asif and James M. Tour and Robert Vajtai and Jun Lou and Douglas S. Galvão and Chandra S. Tiwary and Pulickel. M. Ajayan},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-ultralow-density-graphene-oxidepolydimethylsiloxane-foams/BC2DC24B3DB5714759FC1EDC71BD9D05},
doi = {DOI: 10.1557/adv.2018. 49},
year = {2018},
date = {2018-01-18},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = { 61-66},
abstract = {Low-density, highly porous graphene/graphene oxide (GO) based-foams have shown high performance in energy absorption applications, even under high compressive deformations. In general, foams are very effective as energy dissipative materials and have been widely used in many areas such as automotive, aerospace and biomedical industries. In the case of graphene-based foams, the good mechanical properties are mainly attributed to the intrinsic graphene and/or GO electronic and mechanical properties. Despite the attractive physical properties of graphene/GO based-foams, their structural and thermal stabilities are still a problem for some applications. For instance, they are easily degraded when placed in flowing solutions, either by the collapsing of their layers or just by structural disintegration into small pieces. Recently, a new and scalable synthetic approach to produce low-density 3D macroscopic GO structure interconnected with polydimethylsiloxane (PDMS) polymeric chains (pGO) was proposed. A controlled amount of PDMS is infused into the freeze-dried foam resulting into a very rigid structure with improved mechanical properties, such as tensile plasticity and toughness. The PDMS wets the graphene oxide sheets and acts like a glue bonding PDMS and GO sheets. In order to obtain further insights on mechanisms behind the enhanced mechanical pGO response we carried out fully atomistic molecular dynamics (MD) simulations. Based on MD results, we build up a structural model that can explain the experimentally observed mechanical behavior.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Woellner, Cristiano F.; Botari, Tiago; Perim, Eric; Galvao, Douglas S.
Mechanical Properties of Schwarzites - A Fully Atomistic Reactive Molecular Dynamics Investigation Online
2018, (preprint arXiv:1801.05639).
@online{Woellner2018d,
title = {Mechanical Properties of Schwarzites - A Fully Atomistic Reactive Molecular Dynamics Investigation},
author = {Cristiano F. Woellner and Tiago Botari and Eric Perim and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.05639},
year = {2018},
date = {2018-01-18},
abstract = {Schwarzites are crystalline, 3D porous structures with stable negative curvature formed of sp2-hybridized carbon atoms. These structures present topologies with tunable porous size and shape and unusual mechanical properties. In this work, we have investigated the mechanical behavior under compressive strains and energy absorption of four different Schwarzites, through reactive molecular dynamics simulations, using the ReaxFF force field as available in the LAMMPS code. We considered two Schwarzites families, the so-called Gyroid and Primitive and two structures from each family. Our results also show they exhibit remarkable resilience under mechanical compression. They can be reduced to half of their original size before structural failure (fracture) occurs.},
note = {preprint arXiv:1801.05639},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Jaques, Y. M.; Manimunda, P.; Nakanishi, Y.; Susarla, S.; Woellner, C. F.; Bhowmick, S.; Asif, S. A. S.; Galvao, D. S.; C. S. Tiwary,; Ajayan, P. M.
Differences in the Mechanical Properties of Monolayer and Multilayer WSe2/MoSe2 Online
2018, (preprint arXiv:1801.05641).
@online{Jaques2018b,
title = {Differences in the Mechanical Properties of Monolayer and Multilayer WSe2/MoSe2},
author = {Y. M. Jaques and P. Manimunda and Y. Nakanishi and S. Susarla and C. F. Woellner and S. Bhowmick and S. A. S. Asif and D. S. Galvao and C. S. Tiwary, and P. M. Ajayan},
url = {https://arxiv.org/abs/1801.05641},
year = {2018},
date = {2018-01-18},
abstract = {Transition metal dichalcogenides are 2D structures with remarkable electronic, chemical, optical and mechanical properties. Monolayer and crystal properties of these structures have been extensively investigated, but a detailed understanding of the properties of their few-layer structures are still missing. In this work we investigated the mechanical differences between monolayer and multilayer WSe2 and MoSe2, through fully atomistic molecular dynamics simulations (MD). It was observed that single layer WSe2/MoSe2 deposited on silicon substrates have larger friction coefficients than 2, 3 and 4 layered structures. For all considered cases it is always easier to peel off and/or to fracture MoSe2 structures. These results suggest that the interactions between first layer and substrate are stronger than interlayer interactions themselves. Similar findings have been reported for other nanomaterials and it has been speculated whether this is a universal-like behavior for 2D layered materials. We have also analyzed fracture patterns. Our results show that fracture is chirality dependent with crack propagation preferentially perpendicular to W(Mo)-Se bonds and faster for zig-zag-like defects.},
note = {preprint arXiv:1801.05641},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
de Sousa, Jose M.; Aguiar, Acrisio L.; Girao, Eduardo C.; Fonseca, Alexandre F.; Antonio G. Sousa Filho,; Galvao, Douglas S.
Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation Online
2018, (preprint arXiv:1801.04292).
@online{deSousa2018e,
title = {Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Jose M. de Sousa and Acrisio L. Aguiar and Eduardo C. Girao and Alexandre F. Fonseca and Antonio G. Sousa Filho, and Douglas S. Galvao
},
url = {https://arxiv.org/abs/1801.04292},
year = {2018},
date = {2018-01-12},
abstract = {Recently, a new 2D carbon allotrope structure, named phagraphene (PG), was proposed. PG has a densely array of penta-hexa-hepta-graphene carbon rings. PG was shown to present low and anisotropic thermal conductivity and it is believed that this anisotropy should be also reflected in its mechanical properties. Although PG mechanical properties have been investigated, a detailed and comprehensive study is still lacking. In the present work we have carried out fully atomistic reactive molecular dynamics simulations using the ReaxFF force field, to investigate the mechanical properties and fracture patterns of PG membranes. The Young's modulus values of the PG membranes were estimated from the stress-strain curves. Our results show that these curves present three distinct regimes: one regime where ripples dominate the structure and mechanical properties of the PG membranes; an elastic regime where the membranes exhibit fully planar configurations; and finally a plastic regime where permanent deformations happened to the PG membrane up to the mechanical failure or fracture.},
note = {preprint arXiv:1801.04292},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Sajadi, Seyed Mohammad; Owuor, Peter Samora; Schara, Steven; Woellner, Cristiano F.; Rodrigues, Varlei; Vajtai, Robert; Lou, Jun; Galvao, Douglas S.; Tiwary, Chandra Sekhar; Ajayan, Pulickel M.
Multi-scale Geometric Design Principles Applied to 3D Printed Schwartizes Journal Article
In: Advanced Materials, vol. 2017, pp. 1704820, 2017.
@article{Sajadi2017,
title = {Multi-scale Geometric Design Principles Applied to 3D Printed Schwartizes},
author = {Seyed Mohammad Sajadi and Peter Samora Owuor and Steven Schara and Cristiano F. Woellner and Varlei Rodrigues and Robert Vajtai and Jun Lou and Douglas S. Galvao and Chandra Sekhar Tiwary and Pulickel M. Ajayan},
url = {http://onlinelibrary.wiley.com/doi/10.1002/adma.201704820/full},
doi = {10.1002/adma.201704820},
year = {2017},
date = {2017-09-14},
journal = {Advanced Materials},
volume = {2017},
pages = {1704820},
abstract = {Schwartzites are 3D porous solids with periodic minimal surfaces having negative Gaussian curvatures and can possess unusual mechanical and electronic properties. The mechanical behavior of primitive and gyroid schwartzite structures across different length scales is investigated after these geometries are 3D printed at centimeter length scales based on molec- ular models. Molecular dynamics and nite elements simulations are used
to gain further understanding on responses of these complex solids under compressive loads and kinetic impact experiments. The results show that these structures hold great promise as high load bearing and impact-resistant materials due to a unique layered deformation mechanism that emerges in these architectures during loading. Easily scalable techniques such as 3D printing can be used for exploring mechanical behavior of various predicted complex geometrical shapes to build innovative engineered materials with tunable properties.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
to gain further understanding on responses of these complex solids under compressive loads and kinetic impact experiments. The results show that these structures hold great promise as high load bearing and impact-resistant materials due to a unique layered deformation mechanism that emerges in these architectures during loading. Easily scalable techniques such as 3D printing can be used for exploring mechanical behavior of various predicted complex geometrical shapes to build innovative engineered materials with tunable properties.
Owuor, Peter Samora; Park, Ok-Kyung; Woellner, Cristiano F; Jalilov, Almaz S; Susarla, Sandhya; Joyner, Jarin; Ozden, Sehmus; Duy, LuongXuan; Villegas Salvatierra, Rodrigo; Vajtai, Robert; Tour, James M; Lou, Jun; Galvao, Douglas S; Tiwary, Chandra S; Ajayan, P M
Lightweight Hexagonal Boron Nitride Foam for CO2 Absorption Journal Article
In: ACS Nano, vol. 11, no. 8, pp. 8944–8952, 2017.
@article{Owuor2017b,
title = {Lightweight Hexagonal Boron Nitride Foam for CO2 Absorption},
author = {Owuor, Peter Samora and Park, Ok-Kyung and Woellner, Cristiano F and Jalilov, Almaz S and Susarla, Sandhya and Joyner, Jarin and Ozden, Sehmus and Duy, LuongXuan and Villegas Salvatierra, Rodrigo and Vajtai, Robert and Tour, James M and Lou, Jun and Galvao, Douglas S and Tiwary, Chandra S and Ajayan, P M},
url = {http://pubs.acs.org/doi/abs/10.1021/acsnano.7b03291},
doi = {10.1021/acsnano.7b03291},
year = {2017},
date = {2017-08-03},
journal = {ACS Nano},
volume = {11},
number = {8},
pages = {8944–8952},
abstract = {Weak van der Waals forces between inert hexagonal boron nitride (h-BN) nanosheets make it easy for them to slide over each other, resulting in an unstable structure in macroscopic dimensions. Creating interconnections between these inert nanosheets can remarkably enhance their mechanical properties. However, controlled design of such interconnections remains a fundamental problem for many applications of h-BN foams. In this work, a scalable in situ freeze-drying synthesis of low-density, lightweight 3D macroscopic structures made of h-BN nanosheets chemically connected by poly(vinyl alcohol) (PVA) molecules via chemical cross-link is demonstrated. Unlike pristine h-BN foam which disintegrates upon handling after freeze-drying, h-BN/PVA foams exhibit stable mechanical integrity in addition to high porosity and large surface area. Fully atomistic simulations are used to understand the interactions between h-BN nanosheets and PVA molecules. In addition, the h-BN/PVA foam is investigated as a possible CO2 absorption and as laser irradiation protection material.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
de Sousa, JM; Aguiar, AL; Girao, EC; Fonseca, Alexandre F; AG Filho, Souza; Galvao, Douglas S
Mechanical Properties and Fracture Patterns of Pentagraphene Membranes Online
2017, (preprint arXiv:1703.03789).
@online{deSousa2017,
title = {Mechanical Properties and Fracture Patterns of Pentagraphene Membranes},
author = {de Sousa, JM and Aguiar, AL and Girao, EC and Fonseca, Alexandre F and AG Filho, Souza and Galvao, Douglas S},
url = {https://arxiv.org/abs/1703.03789},
year = {2017},
date = {2017-03-10},
abstract = {Recently, a new two-dimensional carbon allotrope called pentagraphene (PG) was
proposed. PG exhibits mechanical and electronic interesting properties, including typical
band gap values of semiconducting materials. PG has a Cairo-tiling-like 2D lattice
of non coplanar pentagons and its mechanical properties have not been yet fully investigated.
In this work, we combined density functional theory (DFT) calculations and
reactive molecular dynamics (MD) simulations to investigate the mechanical properties
and fracture patterns of PG membranes under tensile strain. We show that PG
membranes can hold up to 20% of strain and that fracture occurs only after substantial
dynamical bond breaking and the formation of 7, 8 and 11 carbon rings and carbon
chains. The stress-strain behavior was observed to follow two regimes, one exhibiting linear elasticity followed by a plastic one, involving carbon atom re-hybridization with
the formation of carbon rings and chains. Our results also show that mechanically
induced structural transitions from PG to graphene is unlikely to occur, in contrast to
what was previously speculated in the literature.},
note = {preprint arXiv:1703.03789},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
proposed. PG exhibits mechanical and electronic interesting properties, including typical
band gap values of semiconducting materials. PG has a Cairo-tiling-like 2D lattice
of non coplanar pentagons and its mechanical properties have not been yet fully investigated.
In this work, we combined density functional theory (DFT) calculations and
reactive molecular dynamics (MD) simulations to investigate the mechanical properties
and fracture patterns of PG membranes under tensile strain. We show that PG
membranes can hold up to 20% of strain and that fracture occurs only after substantial
dynamical bond breaking and the formation of 7, 8 and 11 carbon rings and carbon
chains. The stress-strain behavior was observed to follow two regimes, one exhibiting linear elasticity followed by a plastic one, involving carbon atom re-hybridization with
the formation of carbon rings and chains. Our results also show that mechanically
induced structural transitions from PG to graphene is unlikely to occur, in contrast to
what was previously speculated in the literature.
Cristiano F Woellner Peter Samora Owuor, Tong Li
High Toughness in Ultralow Density Graphene Oxide Foam Journal Article
In: Advanced Materials Interfaces, vol. 4, no. 10, pp. 1700030, 2017.
@article{Owuor2017,
title = {High Toughness in Ultralow Density Graphene Oxide Foam},
author = {Peter Samora Owuor, Cristiano F Woellner, Tong Li, Soumya Vinod, Sehmus Ozden, Suppanat Kosolwattana, Sanjit Bhowmick, Luong Xuan Duy, Rodrigo V Salvatierra, Bingqing Wei, Syed AS Asif, James M Tour, Robert Vajtai, Jun Lou, Douglas S Galvão, Chandra Sekhar Tiwary, Pulickel Ajayan},
url = {http://onlinelibrary.wiley.com/doi/10.1002/admi.201700030/abstract },
doi = {10.1002/admi.201700030},
year = {2017},
date = {2017-03-01},
journal = {Advanced Materials Interfaces},
volume = {4},
number = {10},
pages = {1700030},
abstract = {Here, the scalable synthesis of low-density 3D macroscopic structure of graphene oxide (GO) interconnected with polydimethylsiloxane (PDMS) is reported. A controlled amount of PDMS is infused into the freeze-dried foam to result into a very rigid structure with improved mechanical properties, such as tensile plasticity and toughness. The PDMS wets the graphene oxide sheets and acts like glue between the 2D sheets. Molecular dynamics simulations are used to further elucidate the mechanisms of the interactions of graphene oxide layers with PDMS. The ability of using the interconnecting graphene oxide foam as an effective oil–water separator and stable insulating behavior to elevated temperatures are further demonstrated. The structural rigidity of the sample is also tested using laser impact and compared with GO foam.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Bizao, Rafael A; Botari, Tiago; Perim, Eric; Pugno, Nicola M; Galvao, Douglas S
Mechanical Properties and Fracture Patterns of Graphene (Graphitic) Nanowiggles Online
2017, (preprint arXiv:1702.01100).
@online{Bizao2017,
title = {Mechanical Properties and Fracture Patterns of Graphene (Graphitic) Nanowiggles},
author = {Bizao, Rafael A and Botari, Tiago and Perim, Eric and Pugno, Nicola M and Galvao, Douglas S},
url = {https://arxiv.org/pdf/1702.01100.pdf},
year = {2017},
date = {2017-02-03},
abstract = {Graphene nanowiggles (GNW) are graphene-based nanostructures
obtained by making alternated regular cuts in pristine graphene nanoribbons.
GNW were recently synthesized and it was demonstrated that
they exhibit tunable electronic and magnetic properties by just varying
their shape. Here, we have investigated the mechanical properties and
fracture patterns of a large number of GNW of different shapes and
sizes using fully atomistic reactive molecular dynamics simulations.
Our results show that the GNW mechanical properties are strongly
dependent on its shape and size and, as a general trend narrow sheets
have larger ultimate strength and Young’s modulus than wide ones.
The estimated Young’s modulus values were found to be in a range of
≈ 100 − 1000 GPa and the ultimate strength in a range of ≈ 20 − 110
GPa, depending on GNW shape. Also, super-ductile behaviour under
strain was observed for some structures.},
note = {preprint arXiv:1702.01100},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
obtained by making alternated regular cuts in pristine graphene nanoribbons.
GNW were recently synthesized and it was demonstrated that
they exhibit tunable electronic and magnetic properties by just varying
their shape. Here, we have investigated the mechanical properties and
fracture patterns of a large number of GNW of different shapes and
sizes using fully atomistic reactive molecular dynamics simulations.
Our results show that the GNW mechanical properties are strongly
dependent on its shape and size and, as a general trend narrow sheets
have larger ultimate strength and Young’s modulus than wide ones.
The estimated Young’s modulus values were found to be in a range of
≈ 100 − 1000 GPa and the ultimate strength in a range of ≈ 20 − 110
GPa, depending on GNW shape. Also, super-ductile behaviour under
strain was observed for some structures.
Peter Samora Owuor Alin Cristian Chipara, Sanjit Bhowmick
Structural Reinforcement through Liquid Encapsulation Journal Article
In: Advanced Materials Interfaces, vol. 4, pp. 1600781, 2017.
@article{Chipara2017,
title = {Structural Reinforcement through Liquid Encapsulation},
author = {Alin Cristian Chipara, Peter Samora Owuor, Sanjit Bhowmick, Gustavo Brunetto, SA Asif, Mircea Chipara, Robert Vajtai, Jun Lou, Douglas S Galvao, Chandra Sekhar Tiwary, Pulickel M Ajayan},
url = {http://onlinelibrary.wiley.com/doi/10.1002/admi.201600781/full},
doi = {10.1002/admi.201600781},
year = {2017},
date = {2017-01-23},
journal = {Advanced Materials Interfaces},
volume = {4},
pages = {1600781},
abstract = {The liquid inside a solid material is one of the most common composite materials in nature. The interface between solid–liquid plays an important role in unique deformation. Here, model systems of two polymers (polydimethylsiloxane–polyvinylidenefluoride) are used to make sphere of solid with liquid inside it.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2019
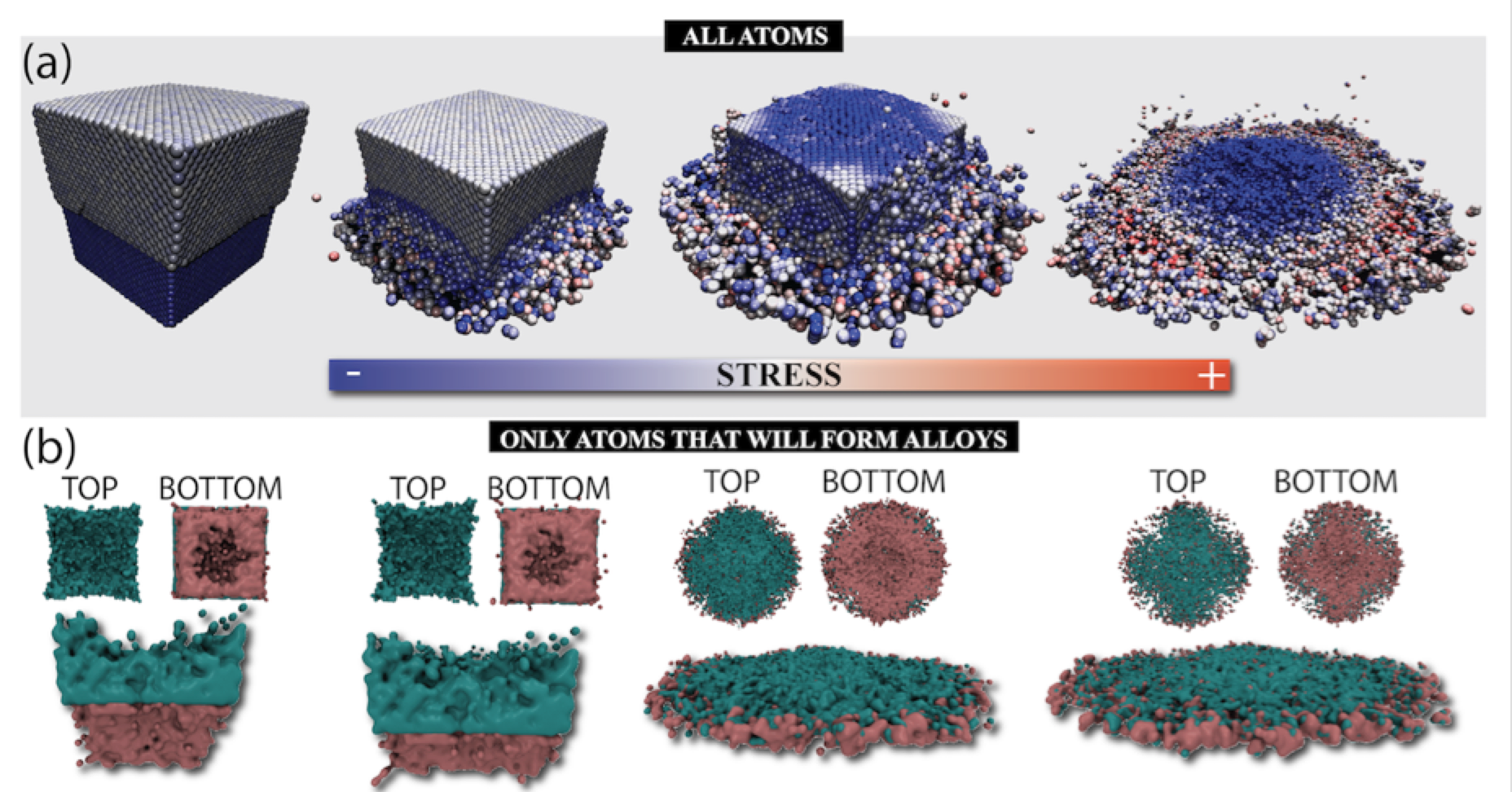
Malviya, Kirtman D; Oliveira, Eliezer F; Autreto, Pedro A S; Ajayan, Pulickel M; Galvao, D S; Tiwary, Candra S; Chattopadhyay, Kumanio
Mixing the immiscible through high-velocity mechanical impacts: an experimental and theoretical study Journal Article
In: Journal of Physics D: Applied Physics, vol. 52, no. 44, pp. 445304, 2019.
Abstract | Links | BibTeX | Tags: Mechanical Properties, Metal, Molecular Dynamics
@article{Malviya2019,
title = {Mixing the immiscible through high-velocity mechanical impacts: an experimental and theoretical study},
author = {Malviya, Kirtman D and Oliveira, Eliezer F and Autreto, Pedro A S and Ajayan, Pulickel M and Galvao, D S and Tiwary, Candra S and Chattopadhyay, Kumanio},
url = {https://iopscience.iop.org/article/10.1088/1361-6463/ab36d1/meta},
doi = {10.1088/1361-6463/ab36d1},
year = {2019},
date = {2019-08-20},
journal = {Journal of Physics D: Applied Physics},
volume = {52},
number = {44},
pages = {445304},
abstract = {In two-component metallic systems, thermodynamic immiscibility leads to phase separation
such as in two-phase eutectic compositional alloys. The limit of the immiscibility of
component elements under non-equilibrium conditions have been explored, but achieving
complete miscibility and formation of single phase microstructures in eutectic alloys would
be unprecedented. Here we report that during low-temperature ball milling that provides high
energy impact, complete mixing of phases can occur in immiscible Ag-Cu eutectic alloys.
From combined theoretical and experimental studies, we show that impact can produce solid
solutions of Ag-Cu nanoparticles of eutectic composition. Our results show that phase
diagrams of low dimensional materials under non-equilibrium conditions remain unexplored
and could lead to new alloy microstructures drastically different from their bulk counterparts.},
keywords = {Mechanical Properties, Metal, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
such as in two-phase eutectic compositional alloys. The limit of the immiscibility of
component elements under non-equilibrium conditions have been explored, but achieving
complete miscibility and formation of single phase microstructures in eutectic alloys would
be unprecedented. Here we report that during low-temperature ball milling that provides high
energy impact, complete mixing of phases can occur in immiscible Ag-Cu eutectic alloys.
From combined theoretical and experimental studies, we show that impact can produce solid
solutions of Ag-Cu nanoparticles of eutectic composition. Our results show that phase
diagrams of low dimensional materials under non-equilibrium conditions remain unexplored
and could lead to new alloy microstructures drastically different from their bulk counterparts.
de Sousa, Jose Moreira; Autreto, Pedro da Silva; Galvao, Douglas Soares
Hydrogenation Dynamics Process of Single-wall Carbon Nanotube Twisted (under review) Journal Article
In: 2019.
BibTeX | Tags: Carbon Nanotubes, Hydrogenation, Mechanical Properties, Molecular Dynamics
@article{deSousa2019d,
title = {Hydrogenation Dynamics Process of Single-wall Carbon Nanotube Twisted (under review)},
author = {de Sousa, Jose Moreira and Autreto, Pedro da Silva and Galvao, Douglas Soares},
year = {2019},
date = {2019-07-15},
keywords = {Carbon Nanotubes, Hydrogenation, Mechanical Properties, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
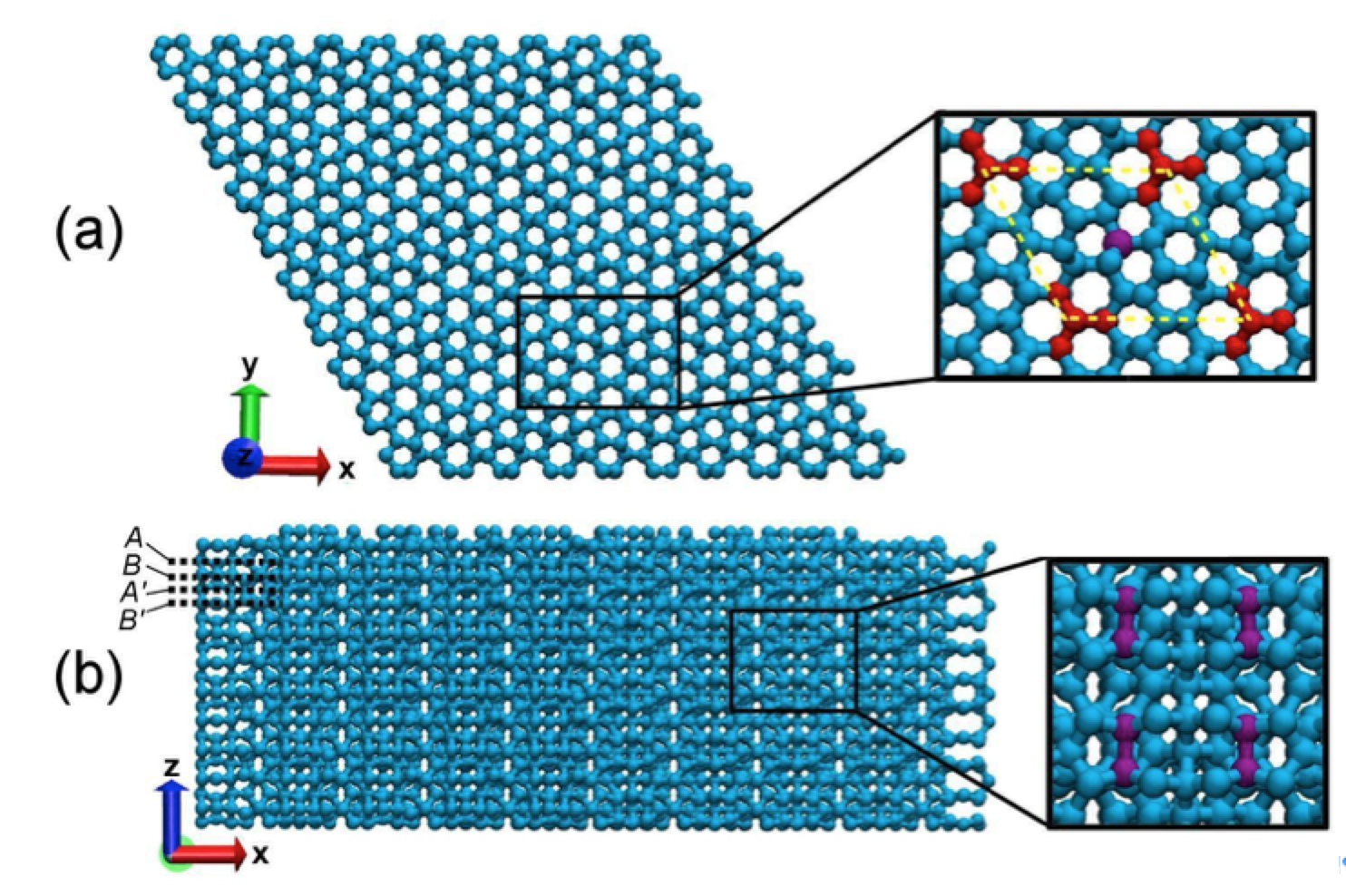
Eliezer F; Autreto Oliveira, Pedro AS; Woellner
On the mechanical properties of protomene: A theoretical investigation Journal Article
In: Computational Materials Science, vol. 161, pp. 190-198, 2019.
Abstract | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, protomene
@article{Oliveira2019c,
title = {On the mechanical properties of protomene: A theoretical investigation},
author = {Oliveira, Eliezer F; Autreto, Pedro AS; Woellner, Cristiano F; Galvao, Douglas S},
year = {2019},
date = {2019-02-07},
journal = {Computational Materials Science},
volume = {161},
pages = {190-198},
abstract = {We report a detailed study through fully atomistic molecular dynamics simulations and DFT calculations on the mechanical properties of protomene. Protomene is a new carbon allotrope composed of a mixture of sp2 and sp3 hybridized states. Our results indicate that protomene presents an anisotropic behavior about tensile deformations. At room temperature, protomene presents an ultimate strength of ~100 GPa and Young's modulus of ~600 GPa, lower than the same for other carbon allotropes. Despite that, protomente presents the highest ultimate strain along the z-direction (~ 24.7%). Our results also show that stretching the protomene along the z-direction or heating it can induce a semiconductor-metallic phase transition, due to a high amount of sp3 bonds that are converted to sp2 ones.},
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, protomene},
pubstate = {published},
tppubtype = {article}
}
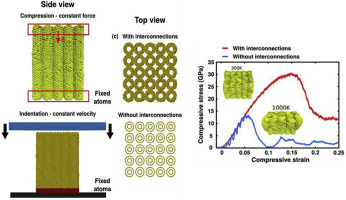
Sanjit; Ozden Bhowmick, Sehmus; Bizão
High temperature quasistatic and dynamic mechanical behavior of interconnected 3D carbon nanotube structures Journal Article
In: Carbon, vol. 142, pp. 291-299, 2019.
Abstract | Links | BibTeX | Tags: CNT, Fracture, Mechanical Properties, Molecular Dynamics
@article{Bhowmick2019,
title = {High temperature quasistatic and dynamic mechanical behavior of interconnected 3D carbon nanotube structures},
author = {Bhowmick, Sanjit; Ozden, Sehmus; Bizão, Rafael A; Machado, Leonardo Dantas; Asif, SA Syed; Pugno, Nicola M; Galvao, Douglas S; Tiwary, Chandra Sekhar; Ajayan, PM},
url = {https://www.sciencedirect.com/science/article/pii/S0008622318308911},
doi = {10.1016/j.carbon.2018.09.075},
year = {2019},
date = {2019-02-01},
journal = {Carbon},
volume = {142},
pages = {291-299},
abstract = {Carbon nanotubes (CNTs) are one of the most appealing materials in recent history for both research and commercial interest because of their outstanding physical, chemical, and electrical properties. This is particularly true for 3D arrangements of CNTs which enable their use in larger scale devices and structures. In this paper, the effect of temperature on the quasistatic and dynamic deformation behavior of 3D CNT structures is presented for the first time. An in situ high-temperature nanomechanical instrument was used inside an SEM at high vacuum to investigate mechanical properties of covalently interconnected CNT porous structures in a wide range of temperature. An irreversible bucking at the base of pillar samples was found as a major mode of deformation at room and elevated temperatures. It has been observed that elastic modulus and critical load to first buckle formation decrease progressively with increasing temperature from 25 °C to 750 °C. To understand fatigue resistance, pillars made from this unique structure were compressed to 100 cycles at room temperature and 750 °C. While the structure showed remarkable resistance to fatigue at room temperature, high temperature significantly lowers fatigue resistance. Molecular dynamics (MD) simulation of compression highlights the critical role played by covalent interconnections which prevent localized bending and improve mechanical properties.},
keywords = {CNT, Fracture, Mechanical Properties, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}

Fonseca, Alexandre F.; Galvao, Douglas S.
Self-tearing and self-peeling of folded graphene nanoribbons Journal Article
In: Carbon, vol. 143, pp. 230-239, 2019.
Abstract | Links | BibTeX | Tags: Fracture, Graphene, Mechanical Properties, Molecular Dynamics
@article{Fonseca2019,
title = {Self-tearing and self-peeling of folded graphene nanoribbons},
author = {Alexandre F. Fonseca and Douglas S. Galvao},
url = {https://www.sciencedirect.com/science/article/pii/S0008622318310431},
doi = {10.1016/j.carbon.2018.11.020},
year = {2019},
date = {2019-01-05},
journal = {Carbon},
volume = {143},
pages = {230-239},
abstract = {A recent experimental study showed that an induced folded flap of graphene can spontaneously drive itself its tearing and peeling off a substrate, thus producing long, micrometer sized, regular trapezoidal-shaped folded graphene nanoribbons. As long as the size of the graphene flaps is above a threshold value, the “tug of war” between the forces of adhesion of graphene-graphene and graphene-substrate, flexural strain of folded region and carbon-carbon (C-C) covalent bonds favor the self-tearing and self-peeling off process. As the detailed information regarding the atomic scale mechanism involved in the process remains not fully understood, we carried out atomistic reactive molecular dynamics simulations to address some features of the process. We show that large thermal fluctuations can prevent the process by increasing the probability of chemical reactions between carbon dangling bonds of adjacent graphene layers. The effects of the strength of attraction between graphene and the substrate on the ribbon growth velocities at the early stages of the phenomenon were also investigated. Structures with initial armchair crack-edges were observed to form more uniform cuts than those having initial zigzag ones. Our results are of importance to help set up new experiments on this phenomenon, especially with samples with nanoscale sized cuts. },
keywords = {Fracture, Graphene, Mechanical Properties, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
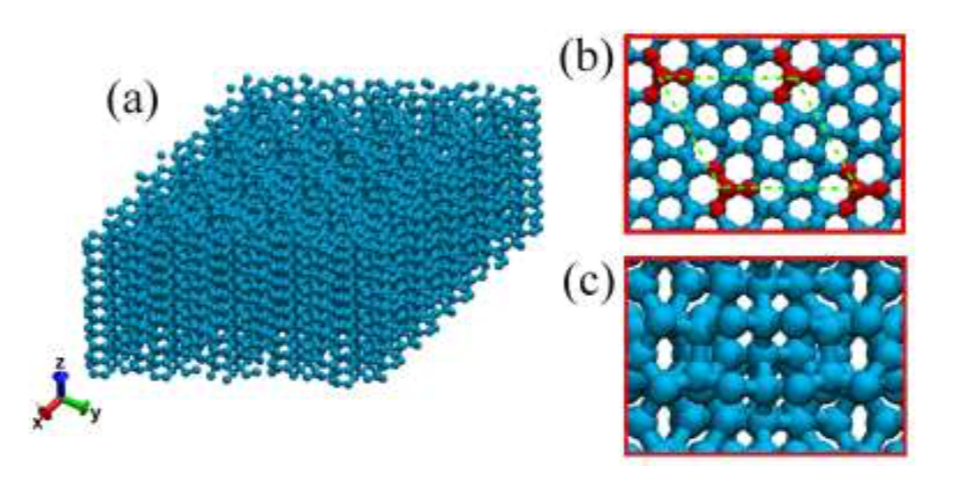
Eliezer F; Autreto Oliveira, Pedro AS; Woellner
Mechanical Properties of Protomene: A Molecular Dynamics Investigation Journal Article
In: MRS Advances, 2019.
Abstract | Links | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, protomene
@article{Oliveira2019,
title = {Mechanical Properties of Protomene: A Molecular Dynamics Investigation},
author = {Oliveira, Eliezer F; Autreto, Pedro AS; Woellner, Cristiano F; Galvao, Douglas S},
url = {www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-protomene-a-molecular-dynamics-investigation/CBAC89BDB5942E3353A5C00BD5D0D9CA},
doi = {10.1557/adv.2018.670},
year = {2019},
date = {2019-01-05},
journal = {MRS Advances},
abstract = {Recently, a new class of carbon allotrope called protomene was proposed. This new structure is composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3 carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now, its mechanical properties have not been investigated. In this work, we have investigated protomene mechanical behavior under tensile strain through fully atomistic reactive molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS code. At room temperature, our results show that the protomene is very stable and the obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical fracture.
},
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, protomene},
pubstate = {published},
tppubtype = {article}
}
2018
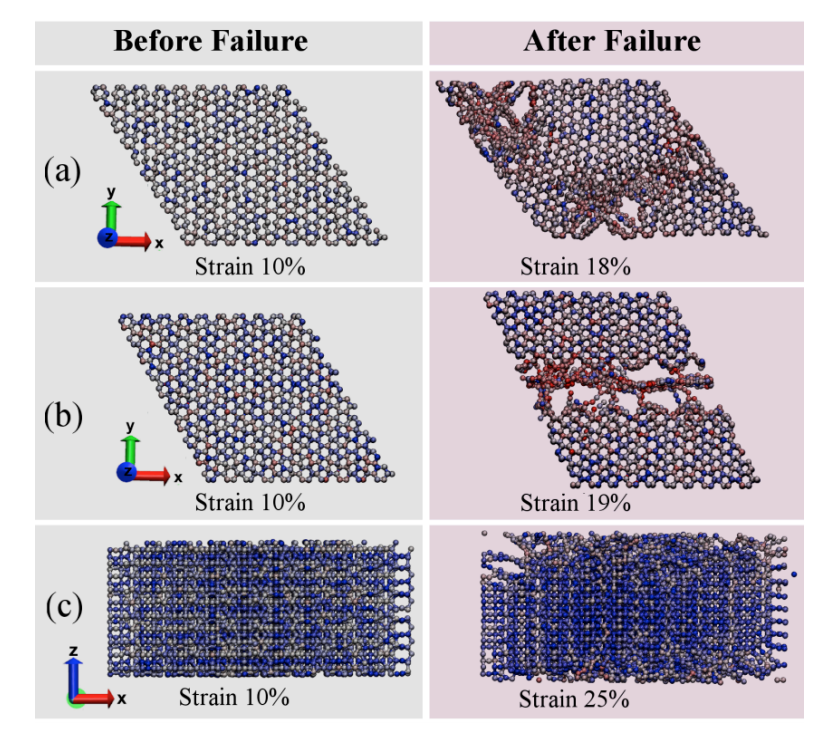
Pedro AS Autreto Eliezer F Oliveira, Cristiano F Woellner
Mechanical Properties of Protomene: A Molecular Dynamics Investigation Online
2018, (preprint arXiv:1810.09924v1 ).
Abstract | Links | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, protomene
@online{Oliveira2018g,
title = {Mechanical Properties of Protomene: A Molecular Dynamics Investigation},
author = {Eliezer F Oliveira, Pedro AS Autreto, Cristiano F Woellner, Douglas S Galvao},
url = {https://arxiv.org/abs/1810.09924},
year = {2018},
date = {2018-10-23},
abstract = {Recently, a new class of carbon allotrope called protomene was proposed. This new structure is
composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3
carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations
have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now,
its mechanical properties have not been investigated. In this work, we have investigated
protomene mechanical behavior under tensile strain through fully atomistic reactive
molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS
code. At room temperature, our results show that the protomene is very stable and the
obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest
ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate
strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical
fracture.},
note = {preprint arXiv:1810.09924v1 },
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, protomene},
pubstate = {published},
tppubtype = {online}
}
composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3
carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations
have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now,
its mechanical properties have not been investigated. In this work, we have investigated
protomene mechanical behavior under tensile strain through fully atomistic reactive
molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS
code. At room temperature, our results show that the protomene is very stable and the
obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest
ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate
strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical
fracture.
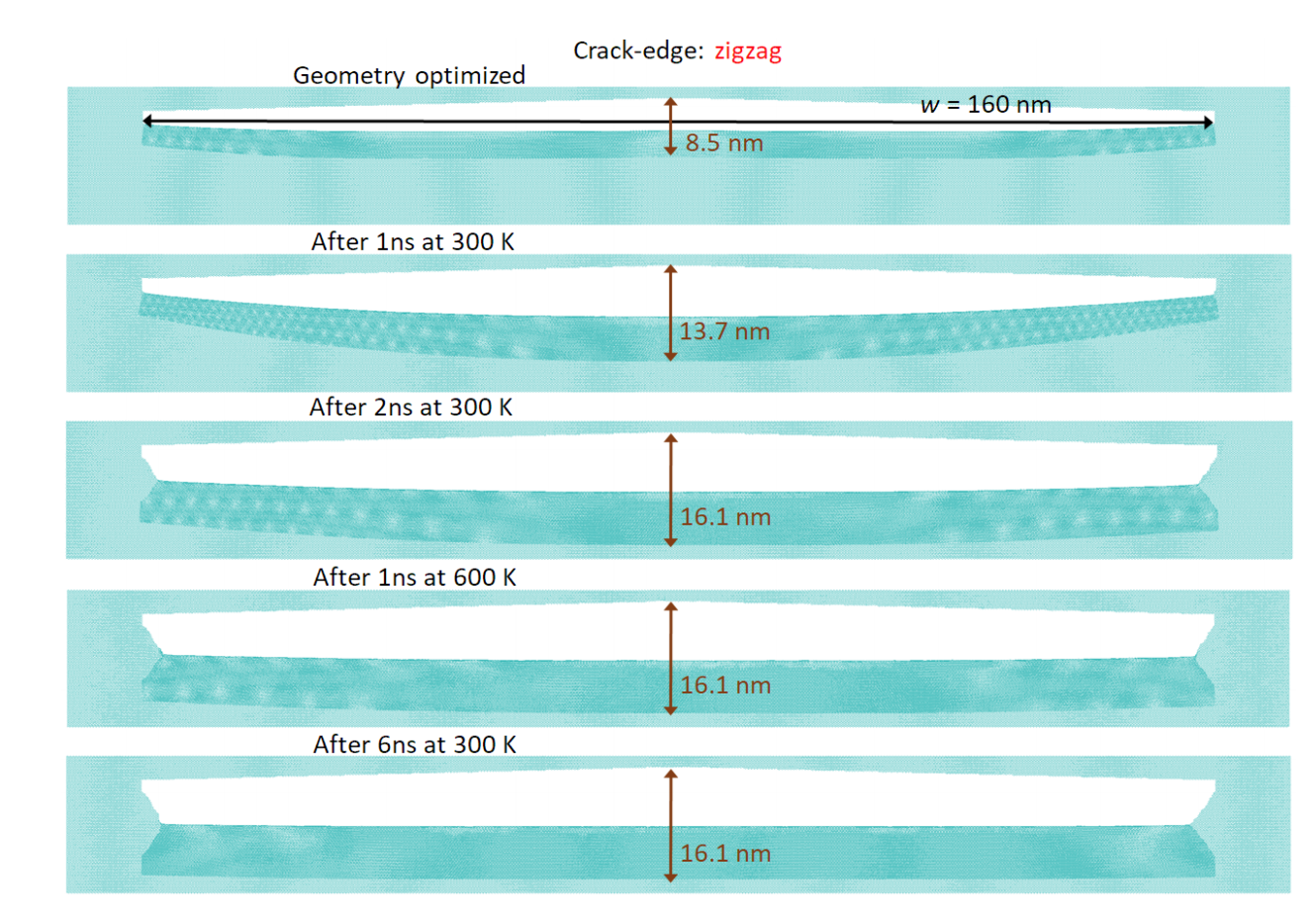
Alexandre F. Fonseca, Douglas S. Galvao
Self-tearing and self-peeling of folded graphene nanoribbons Online
2018, (preprint arXiv:1808.08872).
Abstract | Links | BibTeX | Tags: Fracture, graphene nanoribbons, Mechanical Properties, Molecular Dynamics
@online{Fonseca2018d,
title = { Self-tearing and self-peeling of folded graphene nanoribbons},
author = {Alexandre F. Fonseca, Douglas S. Galvao
},
url = {https://arxiv.org/abs/1808.08872},
year = {2018},
date = {2018-08-27},
abstract = {A recent experimental study showed that an induced folded flap of graphene can spontaneously drive itself its tearing and peeling off a substrate, thus producing long, micrometer sized, regular trapezoidal-shaped folded graphene nanoribbons. As long as the size of the graphene flaps is above a threshold value, the 'tug of war' between the forces of adhesion of graphene-graphene and graphene-substrate, flexural strain of folded region and carbon-carbon (C-C) covalent bonds favor the self-tearing and self-peeling off process. As the detailed information regarding the atomic scale mechanism involved in the process remains not fully understood, we carried out atomistic reactive molecular dynamics simulations to address some features of the process. We show that large thermal fluctuations can prevent the process by increasing the probability of chemical reactions between carbon dangling bonds of adjacent graphene layers. The effects of the strength of attraction between graphene and the substrate on the ribbon growth velocities at the early stages of the phenomenon were also investigated. Structures with initial armchair crack-edges were observed to form more uniform cuts than those having initial zigzag ones. Our results are of importance to help set up new experiments on this phenomenon, especially with samples with nanoscale sized cuts.},
note = {preprint arXiv:1808.08872},
keywords = {Fracture, graphene nanoribbons, Mechanical Properties, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}
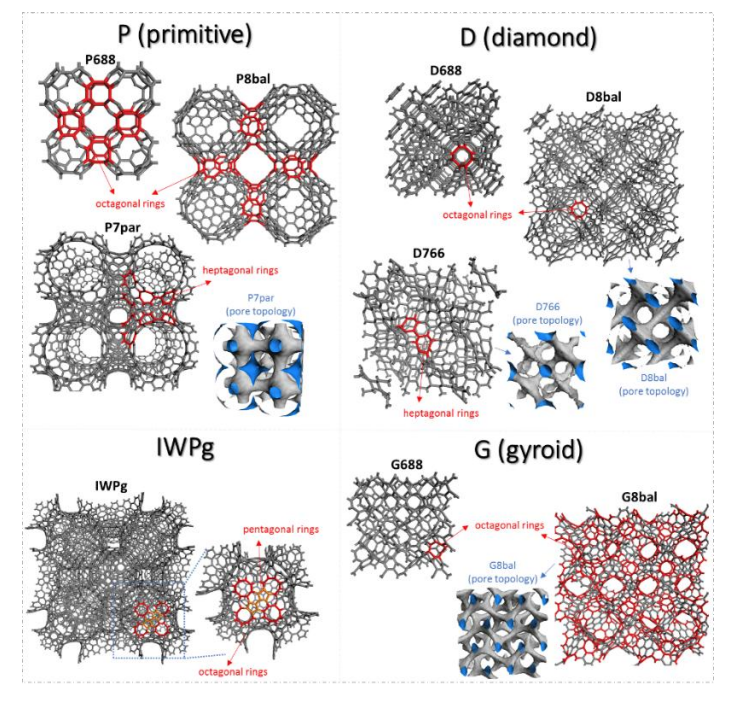
Borges, Daiane Damasceno; Galvao, Douglas S.
Schwarzites for Natural Gas Storage: A Grand-Canonical Monte Carlo Study Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 115-120, 2018.
Abstract | Links | BibTeX | Tags: Gas Storage, Mechanical Properties, Molecular Dynamics, Monte Carlo, Schwarzites
@article{Borges2018d,
title = {Schwarzites for Natural Gas Storage: A Grand-Canonical Monte Carlo Study },
author = {Daiane Damasceno Borges and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/schwarzites-for-natural-gas-storage-a-grandcanonical-monte-carlo-study/2DF8D601AF8EF04BBAC5CCCBEFA8339E},
doi = {https://doi.org/10.1557/adv.2018.190},
year = {2018},
date = {2018-02-13},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = {115-120},
abstract = {he 3D porous carbon-based structures called Schwarzites have been recently a subject of renewed interest due to the possibility of being synthesized in the near future. These structures exhibit negatively curvature topologies with tuneable porous sizes and shapes, which make them natural candidates for applications such as CO2 capture, gas storage and separation. Nevertheless, the adsorption properties of these materials have not been fully investigated. Following this motivation, we have carried out Grand-Canonical Monte Carlo simulations to study the adsorption of small molecules such as CO2, CO, CH4, N2 and H2, in a series of Schwarzites structures. Here, we present our preliminary results on natural gas adsorptive capacity in association with analyses of the guest-host interaction strengths. Our results show that Schwarzites P7par, P8bal and IWPg are the most promising structures with very high CO2 and CH4 adsorption capacity and low saturation pressure (<1bar) at ambient temperature. The P688 is interesting for H2 storage due to its exceptional high H2 adsorption enthalpy value of -19kJ/mol.},
keywords = {Gas Storage, Mechanical Properties, Molecular Dynamics, Monte Carlo, Schwarzites},
pubstate = {published},
tppubtype = {article}
}
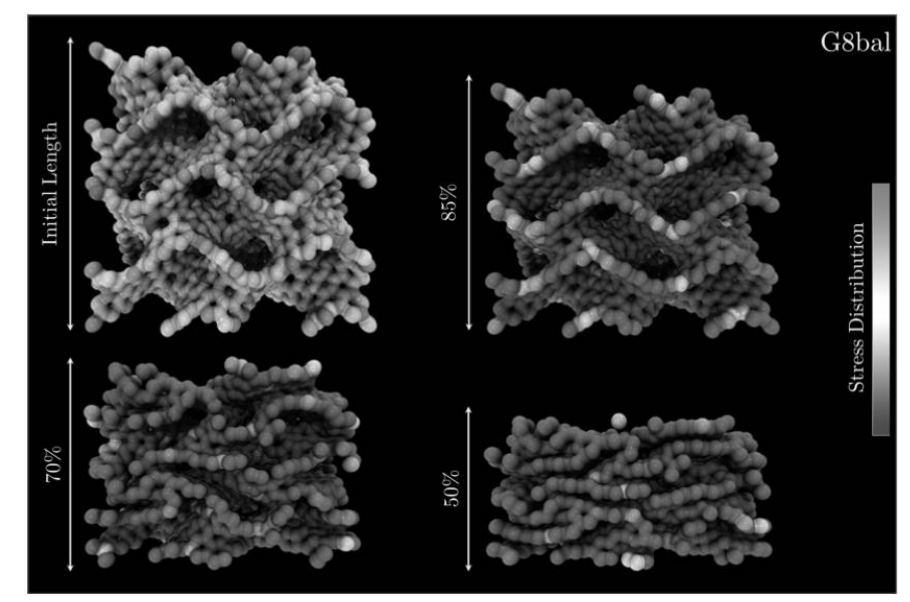
Woellner, Cristiano F.; Botari, Tiago; Perim, Eric; Galvao, Douglas S.
Mechanical Properties of Schwarzites - A Fully Atomistic Reactive Molecular Dynamics Investigation Journal Article
In: MRS Advances, pp. 1-6, 2018.
Abstract | Links | BibTeX | Tags: Mechanical Properties, Molecular Dynamics, Schwarzites
@article{Woellner2018b,
title = {Mechanical Properties of Schwarzites - A Fully Atomistic Reactive Molecular Dynamics Investigation},
author = {Cristiano F. Woellner and Tiago Botari and Eric Perim and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-schwarzites-a-fully-atomistic-reactive-molecular-dynamics-investigation/012AF477491A46541A052C944E4E4834},
doi = { https://doi.org/10.1557/adv.2018.124},
year = {2018},
date = {2018-01-29},
journal = {MRS Advances},
pages = {1-6},
abstract = {Schwarzites are crystalline, 3D porous structures with a stable negative curvature formed of sp2-hybridized carbon atoms. These structures present topologies with tunable porous size and shape and unusual mechanical properties. In this work, we have investigated the mechanical behavior under compressive strain and energy absorption of four different Schwarzites. We considered two Schwarzites families, the so-called Gyroid and Primitive and two structures from each family. We carried out reactive molecular dynamics simulations, using the ReaxFF force field as available in the LAMMPS code. Our results also show they exhibit remarkable resilience under mechanical compression. They can be reduced to half of their original size before structural failure (fracture) occurs.},
keywords = {Mechanical Properties, Molecular Dynamics, Schwarzites},
pubstate = {published},
tppubtype = {article}
}
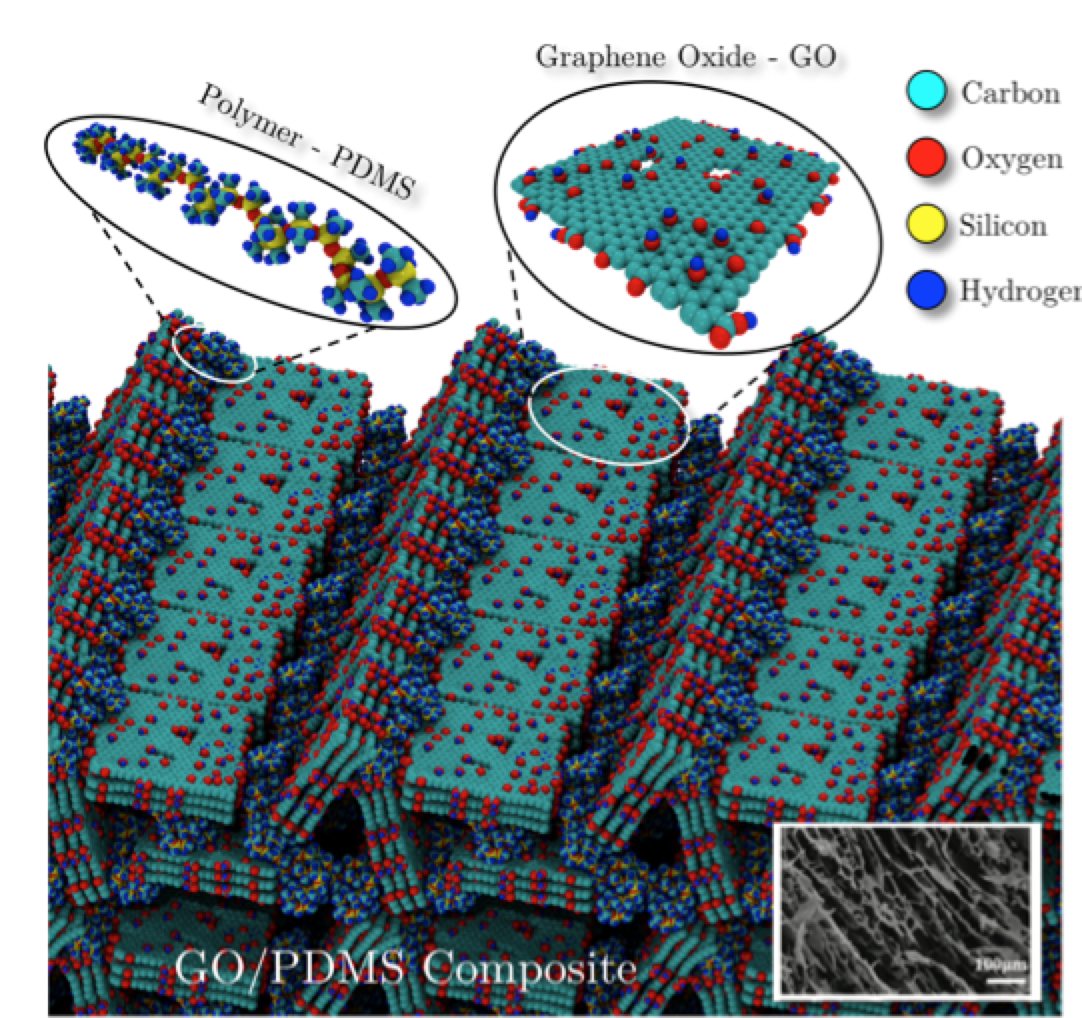
Woellner, Cristiano F.; Owuor, Peter S.; Li, Tong; Vinod, Soumya; Ozden, Sehmus; Kosolwattana, Suppanat; Bhowmick, Sanjit; Duy, Luong X.; Salvatierra, Rodrigo V.; Wei, Bingqing; Asif, Syed A. S.; Tour, James M.; Vajtai, Robert; Lou, Jun; Galvão, Douglas S.; Tiwary, Chandra S.; Ajayan, Pulickel. M.
Mechanical Properties of Ultralow Density Graphene Oxide/Polydimethylsiloxane Foams Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 61-66, 2018.
Abstract | Links | BibTeX | Tags: foams, Mechanical Properties, Molecular Dynamics
@article{Woellner2018c,
title = {Mechanical Properties of Ultralow Density Graphene Oxide/Polydimethylsiloxane Foams},
author = {Cristiano F. Woellner and Peter S. Owuor and Tong Li and Soumya Vinod and Sehmus Ozden and Suppanat Kosolwattana and Sanjit Bhowmick and Luong X. Duy and Rodrigo V. Salvatierra and Bingqing Wei and Syed A. S. Asif and James M. Tour and Robert Vajtai and Jun Lou and Douglas S. Galvão and Chandra S. Tiwary and Pulickel. M. Ajayan},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-ultralow-density-graphene-oxidepolydimethylsiloxane-foams/BC2DC24B3DB5714759FC1EDC71BD9D05},
doi = {DOI: 10.1557/adv.2018. 49},
year = {2018},
date = {2018-01-18},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = { 61-66},
abstract = {Low-density, highly porous graphene/graphene oxide (GO) based-foams have shown high performance in energy absorption applications, even under high compressive deformations. In general, foams are very effective as energy dissipative materials and have been widely used in many areas such as automotive, aerospace and biomedical industries. In the case of graphene-based foams, the good mechanical properties are mainly attributed to the intrinsic graphene and/or GO electronic and mechanical properties. Despite the attractive physical properties of graphene/GO based-foams, their structural and thermal stabilities are still a problem for some applications. For instance, they are easily degraded when placed in flowing solutions, either by the collapsing of their layers or just by structural disintegration into small pieces. Recently, a new and scalable synthetic approach to produce low-density 3D macroscopic GO structure interconnected with polydimethylsiloxane (PDMS) polymeric chains (pGO) was proposed. A controlled amount of PDMS is infused into the freeze-dried foam resulting into a very rigid structure with improved mechanical properties, such as tensile plasticity and toughness. The PDMS wets the graphene oxide sheets and acts like a glue bonding PDMS and GO sheets. In order to obtain further insights on mechanisms behind the enhanced mechanical pGO response we carried out fully atomistic molecular dynamics (MD) simulations. Based on MD results, we build up a structural model that can explain the experimentally observed mechanical behavior.},
keywords = {foams, Mechanical Properties, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
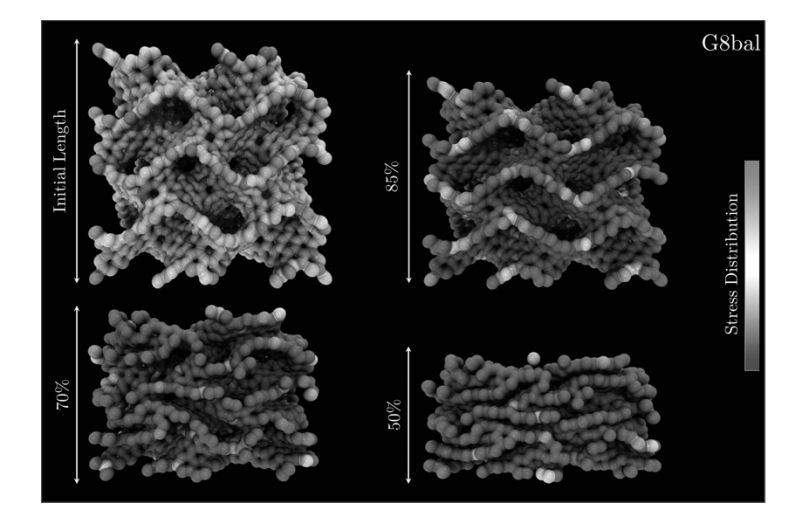
Woellner, Cristiano F.; Botari, Tiago; Perim, Eric; Galvao, Douglas S.
Mechanical Properties of Schwarzites - A Fully Atomistic Reactive Molecular Dynamics Investigation Online
2018, (preprint arXiv:1801.05639).
Abstract | Links | BibTeX | Tags: Mechanical Properties, Molecular Dynamics, Schwarzites
@online{Woellner2018d,
title = {Mechanical Properties of Schwarzites - A Fully Atomistic Reactive Molecular Dynamics Investigation},
author = {Cristiano F. Woellner and Tiago Botari and Eric Perim and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.05639},
year = {2018},
date = {2018-01-18},
abstract = {Schwarzites are crystalline, 3D porous structures with stable negative curvature formed of sp2-hybridized carbon atoms. These structures present topologies with tunable porous size and shape and unusual mechanical properties. In this work, we have investigated the mechanical behavior under compressive strains and energy absorption of four different Schwarzites, through reactive molecular dynamics simulations, using the ReaxFF force field as available in the LAMMPS code. We considered two Schwarzites families, the so-called Gyroid and Primitive and two structures from each family. Our results also show they exhibit remarkable resilience under mechanical compression. They can be reduced to half of their original size before structural failure (fracture) occurs.},
note = {preprint arXiv:1801.05639},
keywords = {Mechanical Properties, Molecular Dynamics, Schwarzites},
pubstate = {published},
tppubtype = {online}
}
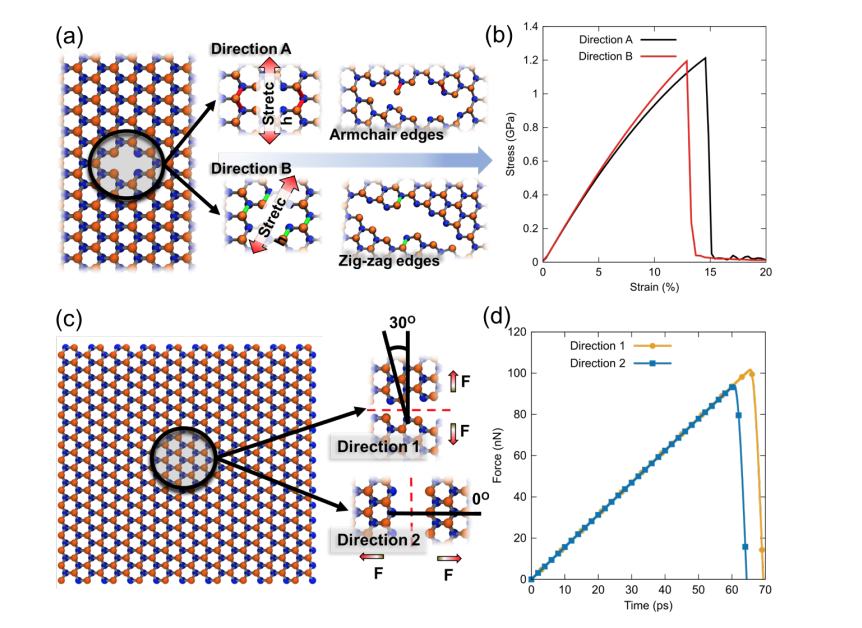
Jaques, Y. M.; Manimunda, P.; Nakanishi, Y.; Susarla, S.; Woellner, C. F.; Bhowmick, S.; Asif, S. A. S.; Galvao, D. S.; C. S. Tiwary,; Ajayan, P. M.
Differences in the Mechanical Properties of Monolayer and Multilayer WSe2/MoSe2 Online
2018, (preprint arXiv:1801.05641).
Abstract | Links | BibTeX | Tags: Chalcogenides, Mechanical Properties, Modeling
@online{Jaques2018b,
title = {Differences in the Mechanical Properties of Monolayer and Multilayer WSe2/MoSe2},
author = {Y. M. Jaques and P. Manimunda and Y. Nakanishi and S. Susarla and C. F. Woellner and S. Bhowmick and S. A. S. Asif and D. S. Galvao and C. S. Tiwary, and P. M. Ajayan},
url = {https://arxiv.org/abs/1801.05641},
year = {2018},
date = {2018-01-18},
abstract = {Transition metal dichalcogenides are 2D structures with remarkable electronic, chemical, optical and mechanical properties. Monolayer and crystal properties of these structures have been extensively investigated, but a detailed understanding of the properties of their few-layer structures are still missing. In this work we investigated the mechanical differences between monolayer and multilayer WSe2 and MoSe2, through fully atomistic molecular dynamics simulations (MD). It was observed that single layer WSe2/MoSe2 deposited on silicon substrates have larger friction coefficients than 2, 3 and 4 layered structures. For all considered cases it is always easier to peel off and/or to fracture MoSe2 structures. These results suggest that the interactions between first layer and substrate are stronger than interlayer interactions themselves. Similar findings have been reported for other nanomaterials and it has been speculated whether this is a universal-like behavior for 2D layered materials. We have also analyzed fracture patterns. Our results show that fracture is chirality dependent with crack propagation preferentially perpendicular to W(Mo)-Se bonds and faster for zig-zag-like defects.},
note = {preprint arXiv:1801.05641},
keywords = {Chalcogenides, Mechanical Properties, Modeling},
pubstate = {published},
tppubtype = {online}
}

de Sousa, Jose M.; Aguiar, Acrisio L.; Girao, Eduardo C.; Fonseca, Alexandre F.; Antonio G. Sousa Filho,; Galvao, Douglas S.
Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation Online
2018, (preprint arXiv:1801.04292).
Abstract | Links | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, phagraphene
@online{deSousa2018e,
title = {Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Jose M. de Sousa and Acrisio L. Aguiar and Eduardo C. Girao and Alexandre F. Fonseca and Antonio G. Sousa Filho, and Douglas S. Galvao
},
url = {https://arxiv.org/abs/1801.04292},
year = {2018},
date = {2018-01-12},
abstract = {Recently, a new 2D carbon allotrope structure, named phagraphene (PG), was proposed. PG has a densely array of penta-hexa-hepta-graphene carbon rings. PG was shown to present low and anisotropic thermal conductivity and it is believed that this anisotropy should be also reflected in its mechanical properties. Although PG mechanical properties have been investigated, a detailed and comprehensive study is still lacking. In the present work we have carried out fully atomistic reactive molecular dynamics simulations using the ReaxFF force field, to investigate the mechanical properties and fracture patterns of PG membranes. The Young's modulus values of the PG membranes were estimated from the stress-strain curves. Our results show that these curves present three distinct regimes: one regime where ripples dominate the structure and mechanical properties of the PG membranes; an elastic regime where the membranes exhibit fully planar configurations; and finally a plastic regime where permanent deformations happened to the PG membrane up to the mechanical failure or fracture.},
note = {preprint arXiv:1801.04292},
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, phagraphene},
pubstate = {published},
tppubtype = {online}
}
2017

Sajadi, Seyed Mohammad; Owuor, Peter Samora; Schara, Steven; Woellner, Cristiano F.; Rodrigues, Varlei; Vajtai, Robert; Lou, Jun; Galvao, Douglas S.; Tiwary, Chandra Sekhar; Ajayan, Pulickel M.
Multi-scale Geometric Design Principles Applied to 3D Printed Schwartizes Journal Article
In: Advanced Materials, vol. 2017, pp. 1704820, 2017.
Abstract | Links | BibTeX | Tags: 3D printing, Mechanical Properties, Molecular Dynamics, Schwarzites
@article{Sajadi2017,
title = {Multi-scale Geometric Design Principles Applied to 3D Printed Schwartizes},
author = {Seyed Mohammad Sajadi and Peter Samora Owuor and Steven Schara and Cristiano F. Woellner and Varlei Rodrigues and Robert Vajtai and Jun Lou and Douglas S. Galvao and Chandra Sekhar Tiwary and Pulickel M. Ajayan},
url = {http://onlinelibrary.wiley.com/doi/10.1002/adma.201704820/full},
doi = {10.1002/adma.201704820},
year = {2017},
date = {2017-09-14},
journal = {Advanced Materials},
volume = {2017},
pages = {1704820},
abstract = {Schwartzites are 3D porous solids with periodic minimal surfaces having negative Gaussian curvatures and can possess unusual mechanical and electronic properties. The mechanical behavior of primitive and gyroid schwartzite structures across different length scales is investigated after these geometries are 3D printed at centimeter length scales based on molec- ular models. Molecular dynamics and nite elements simulations are used
to gain further understanding on responses of these complex solids under compressive loads and kinetic impact experiments. The results show that these structures hold great promise as high load bearing and impact-resistant materials due to a unique layered deformation mechanism that emerges in these architectures during loading. Easily scalable techniques such as 3D printing can be used for exploring mechanical behavior of various predicted complex geometrical shapes to build innovative engineered materials with tunable properties.},
keywords = {3D printing, Mechanical Properties, Molecular Dynamics, Schwarzites},
pubstate = {published},
tppubtype = {article}
}
to gain further understanding on responses of these complex solids under compressive loads and kinetic impact experiments. The results show that these structures hold great promise as high load bearing and impact-resistant materials due to a unique layered deformation mechanism that emerges in these architectures during loading. Easily scalable techniques such as 3D printing can be used for exploring mechanical behavior of various predicted complex geometrical shapes to build innovative engineered materials with tunable properties.
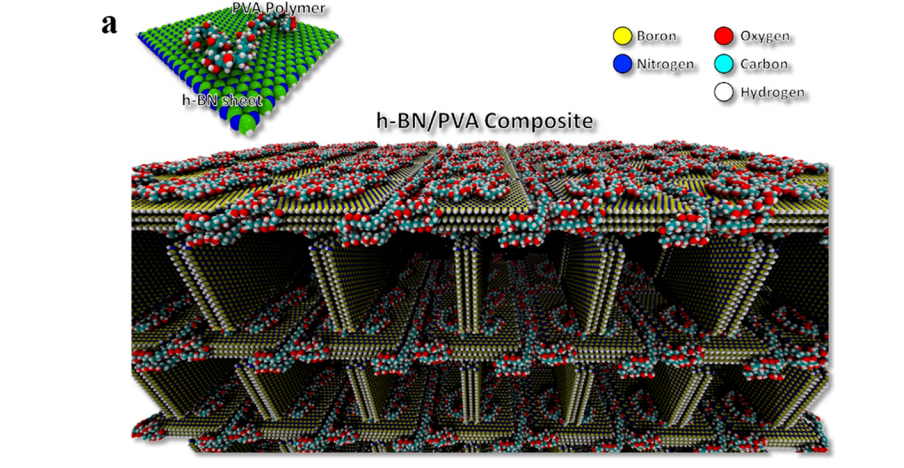
Owuor, Peter Samora; Park, Ok-Kyung; Woellner, Cristiano F; Jalilov, Almaz S; Susarla, Sandhya; Joyner, Jarin; Ozden, Sehmus; Duy, LuongXuan; Villegas Salvatierra, Rodrigo; Vajtai, Robert; Tour, James M; Lou, Jun; Galvao, Douglas S; Tiwary, Chandra S; Ajayan, P M
Lightweight Hexagonal Boron Nitride Foam for CO2 Absorption Journal Article
In: ACS Nano, vol. 11, no. 8, pp. 8944–8952, 2017.
Abstract | Links | BibTeX | Tags: foams, Mechanical Properties, Molecular Dynamics
@article{Owuor2017b,
title = {Lightweight Hexagonal Boron Nitride Foam for CO2 Absorption},
author = {Owuor, Peter Samora and Park, Ok-Kyung and Woellner, Cristiano F and Jalilov, Almaz S and Susarla, Sandhya and Joyner, Jarin and Ozden, Sehmus and Duy, LuongXuan and Villegas Salvatierra, Rodrigo and Vajtai, Robert and Tour, James M and Lou, Jun and Galvao, Douglas S and Tiwary, Chandra S and Ajayan, P M},
url = {http://pubs.acs.org/doi/abs/10.1021/acsnano.7b03291},
doi = {10.1021/acsnano.7b03291},
year = {2017},
date = {2017-08-03},
journal = {ACS Nano},
volume = {11},
number = {8},
pages = {8944–8952},
abstract = {Weak van der Waals forces between inert hexagonal boron nitride (h-BN) nanosheets make it easy for them to slide over each other, resulting in an unstable structure in macroscopic dimensions. Creating interconnections between these inert nanosheets can remarkably enhance their mechanical properties. However, controlled design of such interconnections remains a fundamental problem for many applications of h-BN foams. In this work, a scalable in situ freeze-drying synthesis of low-density, lightweight 3D macroscopic structures made of h-BN nanosheets chemically connected by poly(vinyl alcohol) (PVA) molecules via chemical cross-link is demonstrated. Unlike pristine h-BN foam which disintegrates upon handling after freeze-drying, h-BN/PVA foams exhibit stable mechanical integrity in addition to high porosity and large surface area. Fully atomistic simulations are used to understand the interactions between h-BN nanosheets and PVA molecules. In addition, the h-BN/PVA foam is investigated as a possible CO2 absorption and as laser irradiation protection material.
},
keywords = {foams, Mechanical Properties, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
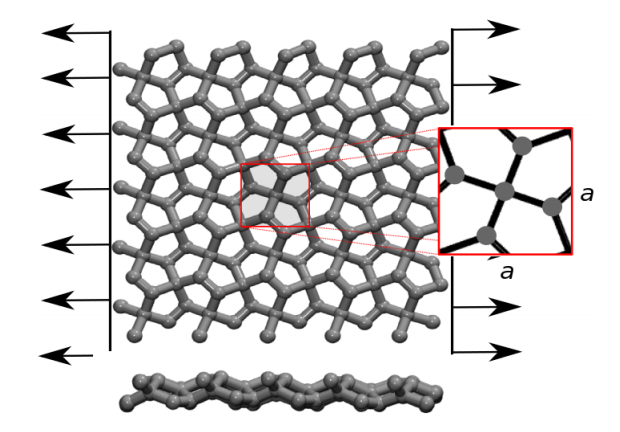
de Sousa, JM; Aguiar, AL; Girao, EC; Fonseca, Alexandre F; AG Filho, Souza; Galvao, Douglas S
Mechanical Properties and Fracture Patterns of Pentagraphene Membranes Online
2017, (preprint arXiv:1703.03789).
Abstract | Links | BibTeX | Tags: DFT, Mechanical Properties, Molecular Dynamics, pentagraphene
@online{deSousa2017,
title = {Mechanical Properties and Fracture Patterns of Pentagraphene Membranes},
author = {de Sousa, JM and Aguiar, AL and Girao, EC and Fonseca, Alexandre F and AG Filho, Souza and Galvao, Douglas S},
url = {https://arxiv.org/abs/1703.03789},
year = {2017},
date = {2017-03-10},
abstract = {Recently, a new two-dimensional carbon allotrope called pentagraphene (PG) was
proposed. PG exhibits mechanical and electronic interesting properties, including typical
band gap values of semiconducting materials. PG has a Cairo-tiling-like 2D lattice
of non coplanar pentagons and its mechanical properties have not been yet fully investigated.
In this work, we combined density functional theory (DFT) calculations and
reactive molecular dynamics (MD) simulations to investigate the mechanical properties
and fracture patterns of PG membranes under tensile strain. We show that PG
membranes can hold up to 20% of strain and that fracture occurs only after substantial
dynamical bond breaking and the formation of 7, 8 and 11 carbon rings and carbon
chains. The stress-strain behavior was observed to follow two regimes, one exhibiting linear elasticity followed by a plastic one, involving carbon atom re-hybridization with
the formation of carbon rings and chains. Our results also show that mechanically
induced structural transitions from PG to graphene is unlikely to occur, in contrast to
what was previously speculated in the literature.},
note = {preprint arXiv:1703.03789},
keywords = {DFT, Mechanical Properties, Molecular Dynamics, pentagraphene},
pubstate = {published},
tppubtype = {online}
}
proposed. PG exhibits mechanical and electronic interesting properties, including typical
band gap values of semiconducting materials. PG has a Cairo-tiling-like 2D lattice
of non coplanar pentagons and its mechanical properties have not been yet fully investigated.
In this work, we combined density functional theory (DFT) calculations and
reactive molecular dynamics (MD) simulations to investigate the mechanical properties
and fracture patterns of PG membranes under tensile strain. We show that PG
membranes can hold up to 20% of strain and that fracture occurs only after substantial
dynamical bond breaking and the formation of 7, 8 and 11 carbon rings and carbon
chains. The stress-strain behavior was observed to follow two regimes, one exhibiting linear elasticity followed by a plastic one, involving carbon atom re-hybridization with
the formation of carbon rings and chains. Our results also show that mechanically
induced structural transitions from PG to graphene is unlikely to occur, in contrast to
what was previously speculated in the literature.
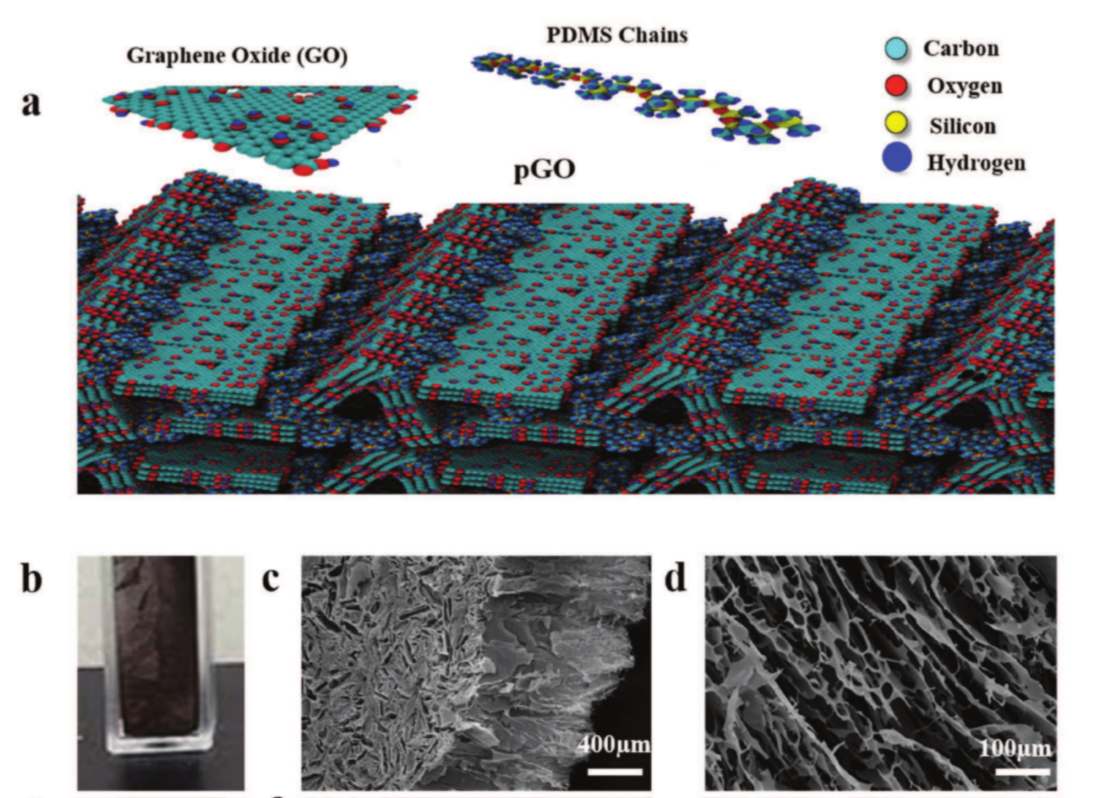
Cristiano F Woellner Peter Samora Owuor, Tong Li
High Toughness in Ultralow Density Graphene Oxide Foam Journal Article
In: Advanced Materials Interfaces, vol. 4, no. 10, pp. 1700030, 2017.
Abstract | Links | BibTeX | Tags: foams, graphene oxide, Mechanical Properties, Molecular Dynamics
@article{Owuor2017,
title = {High Toughness in Ultralow Density Graphene Oxide Foam},
author = {Peter Samora Owuor, Cristiano F Woellner, Tong Li, Soumya Vinod, Sehmus Ozden, Suppanat Kosolwattana, Sanjit Bhowmick, Luong Xuan Duy, Rodrigo V Salvatierra, Bingqing Wei, Syed AS Asif, James M Tour, Robert Vajtai, Jun Lou, Douglas S Galvão, Chandra Sekhar Tiwary, Pulickel Ajayan},
url = {http://onlinelibrary.wiley.com/doi/10.1002/admi.201700030/abstract },
doi = {10.1002/admi.201700030},
year = {2017},
date = {2017-03-01},
journal = {Advanced Materials Interfaces},
volume = {4},
number = {10},
pages = {1700030},
abstract = {Here, the scalable synthesis of low-density 3D macroscopic structure of graphene oxide (GO) interconnected with polydimethylsiloxane (PDMS) is reported. A controlled amount of PDMS is infused into the freeze-dried foam to result into a very rigid structure with improved mechanical properties, such as tensile plasticity and toughness. The PDMS wets the graphene oxide sheets and acts like glue between the 2D sheets. Molecular dynamics simulations are used to further elucidate the mechanisms of the interactions of graphene oxide layers with PDMS. The ability of using the interconnecting graphene oxide foam as an effective oil–water separator and stable insulating behavior to elevated temperatures are further demonstrated. The structural rigidity of the sample is also tested using laser impact and compared with GO foam.},
keywords = {foams, graphene oxide, Mechanical Properties, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
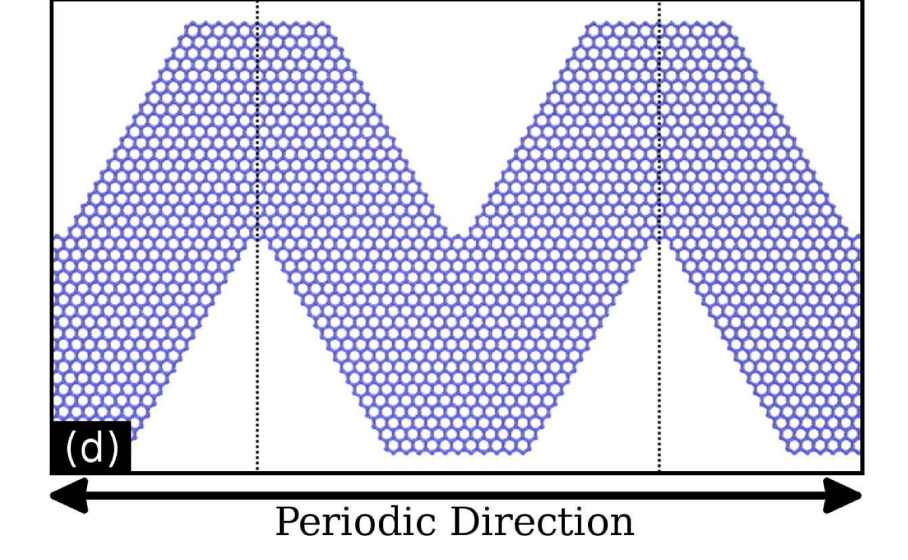
Bizao, Rafael A; Botari, Tiago; Perim, Eric; Pugno, Nicola M; Galvao, Douglas S
Mechanical Properties and Fracture Patterns of Graphene (Graphitic) Nanowiggles Online
2017, (preprint arXiv:1702.01100).
Abstract | Links | BibTeX | Tags: Graphene, Mechanical Properties, Molecular Dynamics, Nanowiggles
@online{Bizao2017,
title = {Mechanical Properties and Fracture Patterns of Graphene (Graphitic) Nanowiggles},
author = {Bizao, Rafael A and Botari, Tiago and Perim, Eric and Pugno, Nicola M and Galvao, Douglas S},
url = {https://arxiv.org/pdf/1702.01100.pdf},
year = {2017},
date = {2017-02-03},
abstract = {Graphene nanowiggles (GNW) are graphene-based nanostructures
obtained by making alternated regular cuts in pristine graphene nanoribbons.
GNW were recently synthesized and it was demonstrated that
they exhibit tunable electronic and magnetic properties by just varying
their shape. Here, we have investigated the mechanical properties and
fracture patterns of a large number of GNW of different shapes and
sizes using fully atomistic reactive molecular dynamics simulations.
Our results show that the GNW mechanical properties are strongly
dependent on its shape and size and, as a general trend narrow sheets
have larger ultimate strength and Young’s modulus than wide ones.
The estimated Young’s modulus values were found to be in a range of
≈ 100 − 1000 GPa and the ultimate strength in a range of ≈ 20 − 110
GPa, depending on GNW shape. Also, super-ductile behaviour under
strain was observed for some structures.},
note = {preprint arXiv:1702.01100},
keywords = {Graphene, Mechanical Properties, Molecular Dynamics, Nanowiggles},
pubstate = {published},
tppubtype = {online}
}
obtained by making alternated regular cuts in pristine graphene nanoribbons.
GNW were recently synthesized and it was demonstrated that
they exhibit tunable electronic and magnetic properties by just varying
their shape. Here, we have investigated the mechanical properties and
fracture patterns of a large number of GNW of different shapes and
sizes using fully atomistic reactive molecular dynamics simulations.
Our results show that the GNW mechanical properties are strongly
dependent on its shape and size and, as a general trend narrow sheets
have larger ultimate strength and Young’s modulus than wide ones.
The estimated Young’s modulus values were found to be in a range of
≈ 100 − 1000 GPa and the ultimate strength in a range of ≈ 20 − 110
GPa, depending on GNW shape. Also, super-ductile behaviour under
strain was observed for some structures.

Peter Samora Owuor Alin Cristian Chipara, Sanjit Bhowmick
Structural Reinforcement through Liquid Encapsulation Journal Article
In: Advanced Materials Interfaces, vol. 4, pp. 1600781, 2017.
Abstract | Links | BibTeX | Tags: Mechanical Properties, Molecular Dynamics, solid-liquid interfaces
@article{Chipara2017,
title = {Structural Reinforcement through Liquid Encapsulation},
author = {Alin Cristian Chipara, Peter Samora Owuor, Sanjit Bhowmick, Gustavo Brunetto, SA Asif, Mircea Chipara, Robert Vajtai, Jun Lou, Douglas S Galvao, Chandra Sekhar Tiwary, Pulickel M Ajayan},
url = {http://onlinelibrary.wiley.com/doi/10.1002/admi.201600781/full},
doi = {10.1002/admi.201600781},
year = {2017},
date = {2017-01-23},
journal = {Advanced Materials Interfaces},
volume = {4},
pages = {1600781},
abstract = {The liquid inside a solid material is one of the most common composite materials in nature. The interface between solid–liquid plays an important role in unique deformation. Here, model systems of two polymers (polydimethylsiloxane–polyvinylidenefluoride) are used to make sphere of solid with liquid inside it.},
keywords = {Mechanical Properties, Molecular Dynamics, solid-liquid interfaces},
pubstate = {published},
tppubtype = {article}
}
2016
T Botari JM de Sousa, E Perim
Mechanical and structural properties of graphene-like carbon nitride sheets Journal Article
In: RSC Advances, vol. 6, no. 80, pp. 76915-76921, 2016.
Abstract | Links | BibTeX | Tags: carbon nitrides sheets, Mechanical Properties, Molecular Dynamics
@article{deSousa2016b,
title = {Mechanical and structural properties of graphene-like carbon nitride sheets},
author = {JM de Sousa, T Botari, E Perim, RA Bizao, Douglas S Galvao},
url = {pubs.rsc.org/en/content/articlehtml/2016/ra/c6ra14273g},
doi = {10.1039/C6RA14273G},
year = {2016},
date = {2016-08-08},
journal = {RSC Advances},
volume = {6},
number = {80},
pages = {76915-76921},
abstract = {Carbon nitride-based nanostructures have attracted special attention (from theory and experiments) due to their remarkable electromechanical properties. In this work we have investigated the mechanical properties of some graphene-like carbon nitride membranes through fully atomistic reactive molecular dynamics simulations. We have analyzed three different structures of these CN families, the so-called graphene-based g-CN, triazine-based g-C3N4 and heptazine-based g-C3N4. The stretching dynamics of these membranes was studied for deformations along their two main axes and at three different temperatures: 10 K, 300 K and 600 K. We show that g-CN membranes have the lowest ultimate fracture strain value, followed by heptazine-based and triazine-based ones, respectively. This behavior can be explained in terms of their differences in density values, topologies and types of chemical bonds. The dependency of the fracture patterns on the stretching directions is also discussed.},
keywords = {carbon nitrides sheets, Mechanical Properties, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
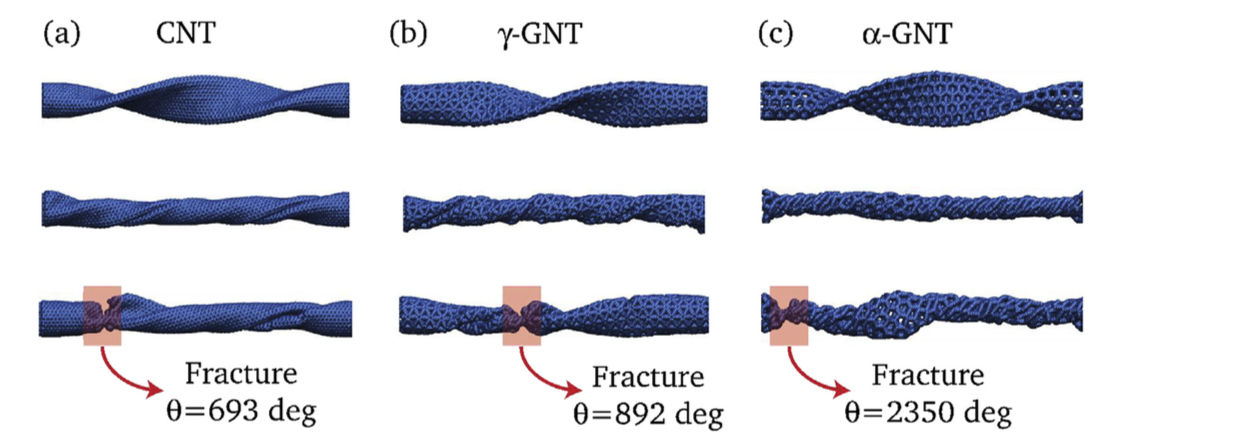
G. Brunetto J.M. de Sousa, V. R. Coluci
Torsional “superplasticity” of graphyne nanotubes Journal Article
In: Carbon, vol. 96, pp. 14-19, 2016.
Abstract | Links | BibTeX | Tags: Fracture, Graphynes, Mechanical Properties, Nanotubes
@article{deSousa2016,
title = {Torsional “superplasticity” of graphyne nanotubes},
author = {J.M. de Sousa, G. Brunetto, V.R. Coluci, D.S. Galvao },
url = {http://www.sciencedirect.com/science/article/pii/S000862231530258X},
doi = { http://dx.doi.org/10.1016/j.carbon.2015.09.039},
year = {2016},
date = {2016-01-01},
journal = {Carbon},
volume = {96},
pages = {14-19},
abstract = {Graphyne is a planar two-dimensional carbon allotrope formed by atoms in sp, sp2, and sp3 hybridized states. Topologically graphyne nanotubes (GNTs) can be considered as cylindrically rolled up graphyne sheets, similarly as carbon nanotubes (CNTs) can be considered rolled up graphene sheets. Due to the presence of single, double, and triple bonds, GNTs exhibit porous sidewalls that can be exploited in many diverse applications. In this work, we investigated the mechanical behavior of GNTs under torsional strains through reactive molecular dynamics simulations. Our results show that GNTs are more flexible than CNTs and exhibit “superplasticit”, with fracture angles that are up to 35 times higher than the ones reported to CNTs. This GNT “superplastic” behavior can be explained in terms of irreversible recon- struction processes (mainly associated with the triple bonds) that occur during torsional strains.},
keywords = {Fracture, Graphynes, Mechanical Properties, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
2015
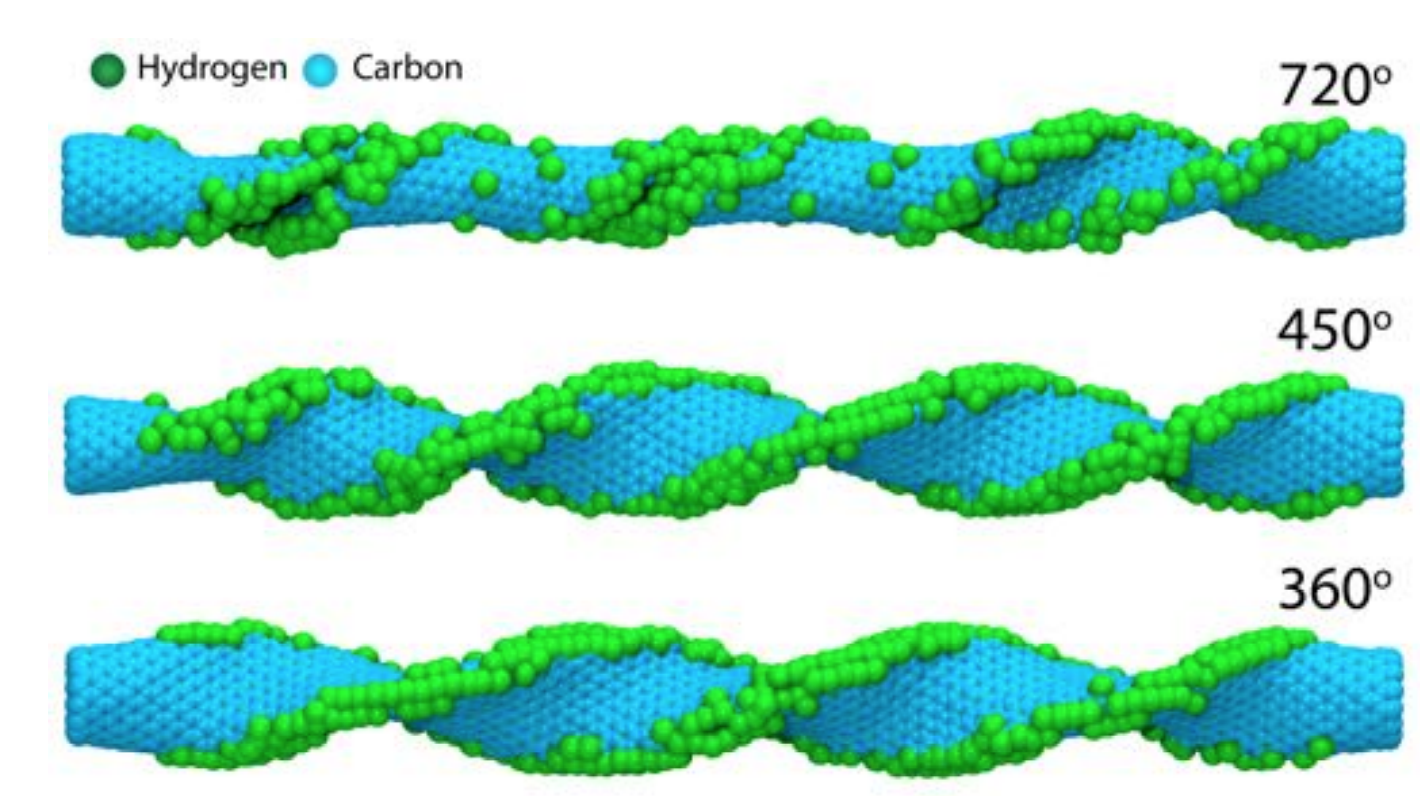
de Sousa, Jose M.; Autreto, Pedro A. S.; Galvao, Douglas S.
Hydrogenation Dynamics of Twisted Carbon Nanotubes Online
2015, (ArXiv preprint).
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, Hydrogenation, Mechanical Properties, Molecular Dynamics
@online{deSousa2015,
title = {Hydrogenation Dynamics of Twisted Carbon Nanotubes},
author = {Jose M. de Sousa and Pedro A. S. Autreto and Douglas S. Galvao},
url = {http://arxiv.org/abs/1510.00265},
year = {2015},
date = {2015-10-01},
abstract = {Carbon Nanotubes (CNTs) are one of the most important materials in nanotechnology. In some of their technological applications (electromechanical oscillators and mechanical actuators for artificial muscles, for instance), it is necessary to subject them to large deformations. Although this frequently happens in air, there are only few studies about the interaction of deformed CNTs with the atmosphere and the dynamics of these processes has not yet been addressed. In this work, we have investigated, through fully atomistic reactive molecular dynamics simulations, the process of hydrogenation of highly twisted CNTs. Our results show that hydrogenation effective ratio is directly related to the tube twist angle values and can lead to twisted tube fractures with well defined patterns (unzip-like). Our results also show that these fracture processes can be exploited to controllably produce graphene nanoribbons.},
note = {ArXiv preprint},
keywords = {Carbon Nanotubes, Hydrogenation, Mechanical Properties, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}
Gustavo Brunetto Jose M. de Sousa, Vitor R. Coluci
Torsional "Superplasticity" of Graphyne Nanotubes Online
2015, (ArXiv reprint of Torsional "Superplasticity" of Graphyne Nanotubes, published in Carbon 96, 14 (2016).).
Abstract | Links | BibTeX | Tags: Allotropes, Graphynes, Mechanical Properties, Nanotubes
@online{deSousa2015b,
title = {Torsional "Superplasticity" of Graphyne Nanotubes},
author = {Jose M. de Sousa, Gustavo Brunetto, Vitor R. Coluci, Douglas S. Galvao},
url = {http://arxiv.org/abs/1509.08746},
year = {2015},
date = {2015-09-29},
abstract = {Graphyne is a planar two-dimensional carbon allotrope formed by atoms in sp, sp2, and sp3 hybridized states. Topologically graphyne nanotubes (GNTs) can be considered as cylindrically rolled up graphyne sheets, similarly as carbon nanotubes (CNTs) can be considered rolled up graphene sheets. Due to the presence of single, double, and triple bonds, GNTs exhibit porous sidewalls that can be exploited in many diverse applications. In this work, we investigated the mechanical behavior of GNTs under torsional strains through reactive molecular dynamics simulations. Our results show that GNTs are more flexible than CNTs and exhibit 'superplasticity', with fracture angles that are up to 35 times higher than the ones reported to CNTs. This GNT 'superplastic' behavior can be explained in terms of irreversible reconstruction processes (mainly associated with the triple bonds) that occur during torsional strains.},
note = {ArXiv reprint of Torsional "Superplasticity" of Graphyne Nanotubes, published in Carbon 96, 14 (2016).},
keywords = {Allotropes, Graphynes, Mechanical Properties, Nanotubes},
pubstate = {published},
tppubtype = {online}
}
Chandra Sekhar Tiwary Dibyendu Chakravarty, Leonardo Dantas Machado
Zirconia-Nanoparticle-Reinforced Morphology-Engineered Graphene-Based Foams Journal Article
In: Advanced Materials, vol. 27, no. 31, pp. 4534–4543, 2015.
Abstract | Links | BibTeX | Tags: Electronic Structure, Mechanical Properties, Mole, Molecular Dynamics, Nanoparticles, Zirconia
@article{Chakravarty2015,
title = {Zirconia-Nanoparticle-Reinforced Morphology-Engineered Graphene-Based Foams},
author = { Dibyendu Chakravarty , Chandra Sekhar Tiwary , Leonardo Dantas Machado ,
Gustavo Brunetto , Soumya Vinod , Ram Manohar Yadav , Douglas S. Galvao ,
Shrikant V. Joshi , Govindan Sundararajan, Pulickel M. Ajayan },
url = {http://onlinelibrary.wiley.com/doi/10.1002/adma.201502409/full},
doi = {10.1002/adma.201502409},
year = {2015},
date = {2015-07-15},
journal = {Advanced Materials},
volume = {27},
number = {31},
pages = {4534–4543},
abstract = {The morphology of graphene-based foams can be engineered by reinforcing them with nanocrystalline zirconia, thus improving their oil-adsorption capacity; This can be observed experimentally and explained theoretically. Low zirconia fractions yield flaky microstructures where zirconia nanoparticles arrest propagating cracks. Higher zirconia concentrations possess a mesh-like interconnected structure where the degree of coiling is dependant on the local zirconia content.},
keywords = {Electronic Structure, Mechanical Properties, Mole, Molecular Dynamics, Nanoparticles, Zirconia},
pubstate = {published},
tppubtype = {article}
}
2014
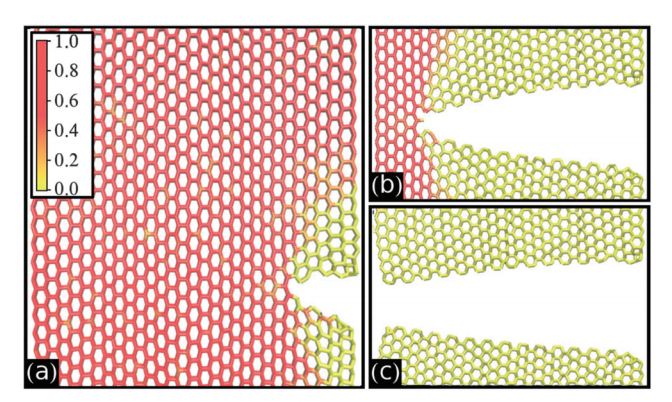
Botari, T; Perim, E; Autreto, PAS; van Duin, ACT; Paupitz, R; Galvao, DS
Mechanical properties and fracture dynamics of silicene membranes Journal Article
In: PHYSICAL CHEMISTRY CHEMICAL PHYSICS, vol. 16, no. 36, pp. 19417–19423, 2014.
Abstract | Links | BibTeX | Tags: Fracture, Germanene, Graphene, Mechanical Properties, Silicene
@article{botari2014mechanical,
title = {Mechanical properties and fracture dynamics of silicene membranes},
author = {Botari, T and Perim, E and Autreto, PAS and van Duin, ACT and Paupitz, R and Galvao, DS},
url = {http://pubs.rsc.org/en/content/articlehtml/2014/cp/c4cp02902j},
year = {2014},
date = {2014-01-01},
journal = {PHYSICAL CHEMISTRY CHEMICAL PHYSICS},
volume = {16},
number = {36},
pages = {19417--19423},
publisher = {ROYAL SOC CHEMISTRY},
abstract = {As graphene has become one of the most important materials, there is renewed interest in other similar structures. One example is silicene, the silicon analogue of graphene. It shares some of the remarkable graphene properties, such as the Dirac cone, but presents some distinct ones, such as a pronounced structural buckling. We have investigated, through density functional based tight-binding (DFTB), as well as reactive molecular dynamics (using ReaxFF), the mechanical properties of suspended single-layer silicene. We calculated the elastic constants, analyzed the fracture patterns and edge reconstructions. We also addressed the stress distributions, unbuckling mechanisms and the fracture dependence on the temperature. We analysed the differences due to distinct edge morphologies, namely zigzag and armchair.},
keywords = {Fracture, Germanene, Graphene, Mechanical Properties, Silicene},
pubstate = {published},
tppubtype = {article}
}

Vinod, Soumya; Tiwary, Chandra Sekhar; da Silva Autreto, Pedro Alves; Taha-Tijerina, Jaime; Ozden, Sehmus; Chipara, Alin Cristian; Vajtai, Robert; Galvao, Douglas S; Narayanan, Tharangattu N; Ajayan, Pulickel M
Low-density three-dimensional foam using self-reinforced hybrid two-dimensional atomic layers Journal Article
In: Nature Communications, vol. 5, 2014.
Links | BibTeX | Tags: foams, Fracture, Graphene, Mechanical Properties, top20
@article{vinod2014low,
title = {Low-density three-dimensional foam using self-reinforced hybrid two-dimensional atomic layers},
author = {Vinod, Soumya and Tiwary, Chandra Sekhar and da Silva Autreto, Pedro Alves and Taha-Tijerina, Jaime and Ozden, Sehmus and Chipara, Alin Cristian and Vajtai, Robert and Galvao, Douglas S and Narayanan, Tharangattu N and Ajayan, Pulickel M},
url = {http://www.nature.com/ncomms/2014/140729/ncomms5541/full/ncomms5541.html},
year = {2014},
date = {2014-01-01},
journal = {Nature Communications},
volume = {5},
publisher = {Nature Publishing Group},
keywords = {foams, Fracture, Graphene, Mechanical Properties, top20},
pubstate = {published},
tppubtype = {article}
}
2013

Perim, E; Autreto, PAS; Paupitz, R; Galvao, DS
Dynamical aspects of the unzipping of multiwalled boron nitride nanotubes Journal Article
In: Physical Chemistry Chemical Physics, vol. 15, no. 44, pp. 19147–19150, 2013.
Abstract | Links | BibTeX | Tags: Boron Nitride, Mechanical Properties, Molecular Dynamics, Unzipping
@article{perim2013dynamical,
title = {Dynamical aspects of the unzipping of multiwalled boron nitride nanotubes},
author = {Perim, E and Autreto, PAS and Paupitz, R and Galvao, DS},
url = {http://pubs.rsc.org/EN/content/articlehtml/2013/cp/c3cp52701h},
year = {2013},
date = {2013-01-01},
journal = {Physical Chemistry Chemical Physics},
volume = {15},
number = {44},
pages = {19147--19150},
publisher = {Royal Society of Chemistry},
abstract = {Boron nitride nanoribbons (BNNRs) exhibit very interesting magnetic properties, which could be very useful in the development of spintronic based devices. One possible route to obtain BNNRs is through the unzipping of boron nitride nanotubes (BNNTs), which have been already experimentally realized. In this work, different aspects of the unzipping process of BNNTs were investigated through fully atomistic molecular dynamics simulations using a classical reactive force field (ReaxFF). We investigated multiwalled BNNTs of different diameters and chiralities. Our results show that chirality plays a very important role in the unzipping process, as well as the interlayer coupling. These combined aspects significantly change the fracturing patterns and several other features of the unzipping processes in comparison to the ones observed for carbon nanotubes. Also, similar to carbon nanotubes, defective BNNTs can create regions of very high curvature which can act as a path to the unzipping process.
},
keywords = {Boron Nitride, Mechanical Properties, Molecular Dynamics, Unzipping},
pubstate = {published},
tppubtype = {article}
}

Perim, Eric; Santos, Ricardo Paupitz; Autreto, Pedro Alves da Silva; Galvao, Douglas S
Fracture Patterns of Boron Nitride Nanotubes Proceedings
Cambridge University Press, vol. 1526, 2013.
Abstract | Links | BibTeX | Tags: Boron Nitride, Fracture, Mechanical Properties, Unzipping
@proceedings{perim2013fracture,
title = {Fracture Patterns of Boron Nitride Nanotubes},
author = {Perim, Eric and Santos, Ricardo Paupitz and Autreto, Pedro Alves da Silva and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8883390&fileId=S1946427413004946},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1526},
pages = {mrsf12--1526},
publisher = {Cambridge University Press},
abstract = {During the last years carbon-based nanostructures (such as, fullerenes, carbon nanotubes and graphene) have been object of intense investigations. The great interest in these nanostructures can be attributed to their remarkable electrical and mechanical properties. Their inorganic equivalent structures do exist and are based on boron nitride (BN) motifs. BN fullerenes, nanotubes and single layers have been already synthesized. Recently, the fracture patterns of single layer graphene and multi-walled carbon nanotubes under stress have been studied by theoretical and experimental methods. In this work we investigated the fracturing process of defective carbon and boron nitride nanotubes under similar stress conditions. We have carried out fully atomistic molecular reactive molecular dynamics simulations using the ReaxFF force field. The similarities and differences between carbon and boron nitride fracture patterns are addressed.},
keywords = {Boron Nitride, Fracture, Mechanical Properties, Unzipping},
pubstate = {published},
tppubtype = {proceedings}
}
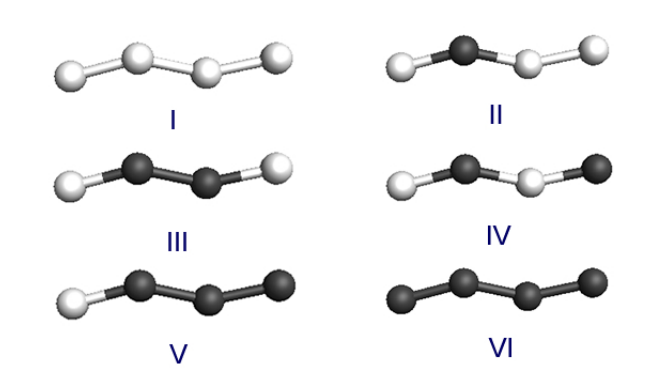
Autreto, Pedro Alves da Silva; Galvao, Douglas S; Artacho, Emilio
Species Fractionation in Atomic Chains from Mechanically Stretched Alloys Journal Article
In: arXiv preprint arXiv:1312.1285, 2013.
Abstract | Links | BibTeX | Tags: Atomic Chains, DFT, Mech, Mechanical Properties, Metallic Nanowires
@article{autreto2013species,
title = {Species Fractionation in Atomic Chains from Mechanically Stretched Alloys},
author = {Autreto, Pedro Alves da Silva and Galvao, Douglas S and Artacho, Emilio},
url = {http://arxiv.org/abs/1312.1285},
year = {2013},
date = {2013-01-01},
journal = {arXiv preprint arXiv:1312.1285},
abstract = {Bettini et al. [Nature Nanotech 1, 182 (2006)] reported the first experimental realization of linear
atomic chains (LACs) composed of different atoms (Au and Ag). Different contents of Au and Ag
were observed in the chains from what found in the bulk alloys, which rises the question of what is the
wire composition if in equilibrium with a bulk alloy. In this work we address the thermodynamic
driving force for species fractionation in LACs under tension, and we present density-functional
theory results for Ag-Au chain alloys. A pronounced stabilization of wires with an alternating
Ag-Au sequence is observed, which could be behind the experimentally observed Au enrichment in
LACs from alloys of high Ag content.},
keywords = {Atomic Chains, DFT, Mech, Mechanical Properties, Metallic Nanowires},
pubstate = {published},
tppubtype = {article}
}
atomic chains (LACs) composed of different atoms (Au and Ag). Different contents of Au and Ag
were observed in the chains from what found in the bulk alloys, which rises the question of what is the
wire composition if in equilibrium with a bulk alloy. In this work we address the thermodynamic
driving force for species fractionation in LACs under tension, and we present density-functional
theory results for Ag-Au chain alloys. A pronounced stabilization of wires with an alternating
Ag-Au sequence is observed, which could be behind the experimentally observed Au enrichment in
LACs from alloys of high Ag content.
2012
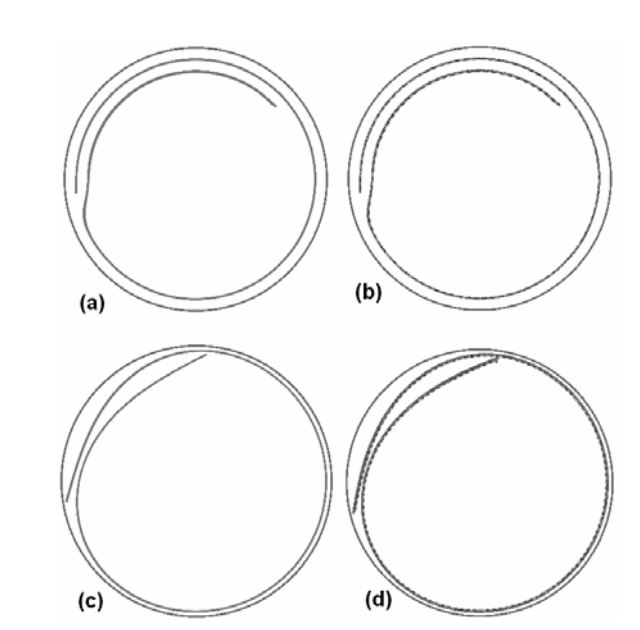
Perim, Eric; Fonseca, Alexandre F; Galvao, Douglas S
When Small is Different: The Case of Membranes Inside Tubes Proceedings
Cambridge University Press, vol. 1451, 2012.
Abstract | Links | BibTeX | Tags: Mechanical Properties, Membranes, Nanoscale Effects, Scrolls
@proceedings{perim2012small,
title = {When Small is Different: The Case of Membranes Inside Tubes},
author = {Perim, Eric and Fonseca, Alexandre F and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8637821&fileId=S1946427412012523},
year = {2012},
date = {2012-01-01},
journal = {MRS Proceedings},
volume = {1451},
pages = {15--20},
publisher = {Cambridge University Press},
abstract = {Recently, classical elasticity theory for thin sheets was used to demonstrate the existence of a universal structural behavior describing the confinement of sheets inside cylindrical tubes. However, this kind of formalism was derived to describe macroscopic systems. A natural question is whether this behavior still holds at nanoscale. In this work, we have investigated through molecular dynamics simulations the structural behavior of graphene and boron nitride single layers confined into nanotubes. Our results show that the class of universality observed at macroscale is no longer observed at nanoscale. The origin of this discrepancy is addressed in terms of the relative importance of forces and energies at macro and nano scales.},
keywords = {Mechanical Properties, Membranes, Nanoscale Effects, Scrolls},
pubstate = {published},
tppubtype = {proceedings}
}
2011
Brunetto, Gustavo; Legoas, Sergio B; Coluci, Vitor R; Lucena, Liacir S; Galvao, Douglas S
Dynamics of Graphene Nanodrums Proceedings
Cambridge University Press, vol. 1284, 2011.
Abstract | Links | BibTeX | Tags: Graphene Membranes, Mechanical Properties, Nanodrum
@proceedings{brunetto2011dynamics,
title = {Dynamics of Graphene Nanodrums},
author = {Brunetto, Gustavo and Legoas, Sergio B and Coluci, Vitor R and Lucena, Liacir S and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8195889&fileId=S1946427411002272},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1284},
pages = {mrsf10--1284},
publisher = {Cambridge University Press},
abstract = {Recently, it was proposed that graphene sheets deposited on silicon oxide can act as impermeable atomic membranes to standard gases, such as helium, argon, and nitrogen. It is assumed that graphene membrane is clamped over the surface due only to van der Waals forces. The leakage mechanism can be experimentally addressed only indirectly. In this work we have carried out molecular dynamics simulations to study this problem. We have considered nano-containers composed of a chamber of silicon oxide filled with gas and sealed by single and multi-layer graphene membranes. The obtained results are in good qualitative agreement with the experimental data. We observed that the graphene membranes remain attached to the substrate for pressure values up to two times the largest value experimentally investigated. We did not observe any gas leakage through the membrane/substrate interface until the critical limit is reached and then a sudden membrane detachment occurs.},
keywords = {Graphene Membranes, Mechanical Properties, Nanodrum},
pubstate = {published},
tppubtype = {proceedings}
}

Autreto, PAS; Lagos, MJ; Sato, F; Bettini, J; Rocha, AR; Rodrigues, V; Ugarte, D; Galvao, DS
Intrinsic Stability of the Smallest Possible Silver Nanotube Journal Article
In: Physical Review Letters, vol. 106, no. 6, pp. 065501, 2011.
Abstract | Links | BibTeX | Tags: DFT, Mechanical Properties, Metallic Nanowires, New Structures, top20
@article{autreto2011intrinsic,
title = {Intrinsic Stability of the Smallest Possible Silver Nanotube},
author = {Autreto, PAS and Lagos, MJ and Sato, F and Bettini, J and Rocha, AR and Rodrigues, V and Ugarte, D and Galvao, DS},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.106.065501},
year = {2011},
date = {2011-01-01},
journal = {Physical Review Letters},
volume = {106},
number = {6},
pages = {065501},
publisher = {American Physical Society},
abstract = {Recently, Lagos et al. [Nature Nanotech. 4, 149 (2009)] reported the discovery of the smallest possible Ag nanotube with a square cross section. Ab initio density functional theory calculations strongly support that the stability of these hollow structures is structurally intrinsic and not the result of contamination by light atoms. We also report the first experimental observation of the theoretically predicted corrugation of the hollow structure. Quantum conductance calculations predict a unique signature of 3.6G0 for this new family of nanotubes.},
keywords = {DFT, Mechanical Properties, Metallic Nanowires, New Structures, top20},
pubstate = {published},
tppubtype = {article}
}
Lagos, Maureen J; Sato, Fernando; Galvao, Douglas S; Ugarte, Daniel
Mechanical deformation of nanoscale metal rods: when size and shape matter Journal Article
In: Physical Review Letters, vol. 106, no. 5, pp. 055501, 2011.
Abstract | Links | BibTeX | Tags: Defects, DFT, Mechanical Properties, Metallic Nanowires
@article{lagos2011mechanical,
title = {Mechanical deformation of nanoscale metal rods: when size and shape matter},
author = {Lagos, Maureen J and Sato, Fernando and Galvao, Douglas S and Ugarte, Daniel},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.106.055501},
year = {2011},
date = {2011-01-01},
journal = {Physical Review Letters},
volume = {106},
number = {5},
pages = {055501},
publisher = {American Physical Society},
abstract = {Face centered cubic metals deform mainly by propagating partial dislocations generating planar fault ribbons. How do metals deform if the size is smaller than the fault ribbons? We studied the elongation of Au and Pt nanorods by in situ electron microscopy and ab initio calculations. Planar fault activation barriers are so low that, for each temperature, a minimal rod size is required to become active for releasing elastic energy. Surface effects dominate deformation energetics; system size and shape determine the preferred fault gliding directions which induce different tensile and compressive behavior.
},
keywords = {Defects, DFT, Mechanical Properties, Metallic Nanowires},
pubstate = {published},
tppubtype = {article}
}

Machado, Leonardo D; Legoas, Sergio B; Soares, Jaqueline S; Shadmi, Nitzan; Jorio, Ado; Joselevich, Ernesto; Galvao, Douglas S
Cambridge University Press, vol. 1284, 2011.
Abstract | Links | BibTeX | Tags: Mechanical Properties, Molecular Dynamics, Serpentines
@proceedings{machado2011formation,
title = {On the formation of carbon nanotube serpentines: insights from multi-million atom molecular dynamics simulation},
author = {Machado, Leonardo D and Legoas, Sergio B and Soares, Jaqueline S and Shadmi, Nitzan and Jorio, Ado and Joselevich, Ernesto and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8194288&fileId=S194642741100220X},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1284},
pages = {mrsf10--1284},
publisher = {Cambridge University Press},
abstract = {In this work we present preliminary results from molecular dynamics simulations for carbon nanotubes serpentine dynamics formation. These S-like nanostructures consist of a series of parallel and straight nanotube segments connected by alternating U-turn shaped curves. Nanotube serpentines were experimentally synthesized and reported in recent years, but up to now no atomistic simulations have been carried out to address the dynamics of formation of these structures. We have carried out fully atomistic molecular dynamics simulations in the framework of classical mechanics with a standard molecular force field. Multi-million atoms structures formed by stepped substrates with a carbon nanotube (about 1 micron in length) placed on top of them have been considered in our simulations. A force is applied to the upper part of the tube during a short period of time and then turned off and the system set free to evolve in time. Our results showed that these conditions are sufficient to form robust serpentines and validate the general features of the ‘falling spaghetti mechanism’ previously proposed to explain their formation.},
keywords = {Mechanical Properties, Molecular Dynamics, Serpentines},
pubstate = {published},
tppubtype = {proceedings}
}
Lagos, MJ; Autreto, PAS; Legoas, SB; Sato, F; Rodrigues, V; Galvao, DS; Ugarte, D
Temperature effects on the occurrence of long interatomic distances in atomic chains formed from stretched gold nanowires Journal Article
In: Nanotechnology, vol. 22, no. 9, pp. 095705, 2011.
Abstract | Links | BibTeX | Tags: Gold, Mechanical Properties, Metallic Nanowires
@article{lagos2011temperature,
title = {Temperature effects on the occurrence of long interatomic distances in atomic chains formed from stretched gold nanowires},
author = {Lagos, MJ and Autreto, PAS and Legoas, SB and Sato, F and Rodrigues, V and Galvao, DS and Ugarte, D},
url = {http://iopscience.iop.org/0957-4484/21/48/485702},
year = {2011},
date = {2011-01-01},
journal = {Nanotechnology},
volume = {22},
number = {9},
pages = {095705},
publisher = {IOP Publishing},
abstract = {We have studied the changes induced by thermal effects in the structural and transport response of Au nanowires generated by mechanical elongation. We have used time-resolved atomic resolution transmission electron microscopy imaging and quantum conductance measurement using a mechanically controllable break junction. Our results showed remarkable differences in the NW evolution for experiments realized at 150 and 300 K, which modifies drastically the conductance response during elongation. Molecular dynamics and electronic transport calculations were used to consistently correlate the observed structural and conductance behavior. These results emphasize that it is essential to take into account the precise atomic arrangement of nanocontacts generated by mechanical stretching to understand electrical transport properties. Also, our study shows that much care must be taken when comparing results obtained in different experimental conditions, mainly different temperatures.
},
keywords = {Gold, Mechanical Properties, Metallic Nanowires},
pubstate = {published},
tppubtype = {article}
}
2010
Martins, BVC; Galvao, DS
Curved graphene nanoribbons: structure and dynamics of carbon nanobelts Journal Article
In: Nanotechnology, vol. 21, no. 7, pp. 075710, 2010.
Abstract | Links | BibTeX | Tags: Folding, Graphene, Mechanical Properties, Nanobelts, NanoRibbons
@article{martins2010curved,
title = {Curved graphene nanoribbons: structure and dynamics of carbon nanobelts},
author = {Martins, BVC and Galvao, DS},
url = {http://iopscience.iop.org/0957-4484/21/7/075710},
year = {2010},
date = {2010-01-01},
journal = {Nanotechnology},
volume = {21},
number = {7},
pages = {075710},
publisher = {IOP Publishing},
abstract = {Carbon nanoribbons (CNRs) are graphene (planar) structures with a large aspect ratio. Carbon nanobelts (CNBs) are small graphene nanoribbons rolled up into spiral-like structures, i.e. carbon nanoscrolls (CNSs) with a large aspect ratio. In this work we investigated the energetics and dynamical aspects of CNBs formed from rolling up CNRs. We have carried out molecular dynamics simulations using reactive empirical bond-order potentials. Our results show that, similarly to CNSs, CNB formation is dominated by two major energy contributions, the increase in the elastic energy due to the bending of the initial planar configuration (decreasing structural stability) and the energetic gain due to van der Waals interactions of the overlapping surface of the rolled layers (increasing structural stability). Beyond a critical diameter value these scrolled structures can be even more stable (in terms of energy) than their equivalent planar configurations. In contrast to CNSs that require energy-assisted processes (sonication, chemical reactions, etc) to be formed, CNBs can be spontaneously formed from low temperature driven processes. Long CNBs (length of ~30.0 nm) tend to exhibit self-folded racket-like conformations with formation dynamics very similar to the one observed for long carbon nanotubes. Shorter CNBs will be more likely to form perfect scrolled structures. Possible synthetic routes to fabricate CNBs from graphene membranes are also addressed.
},
keywords = {Folding, Graphene, Mechanical Properties, Nanobelts, NanoRibbons},
pubstate = {published},
tppubtype = {article}
}
Lagos, MJ; Sato, F; Autreto, PAS; Galvao, DS; Rodrigues, V; Ugarte, D
Temperature effects on the atomic arrangement and conductance of atomic-size gold nanowires generated by mechanical stretching Journal Article
In: Nanotechnology, vol. 21, no. 48, pp. 485702, 2010.
Abstract | Links | BibTeX | Tags: DFT, Mechanical Properties, Metallic Nanowires, Quantum Transport, TEM
@article{lagos2010temperature,
title = {Temperature effects on the atomic arrangement and conductance of atomic-size gold nanowires generated by mechanical stretching},
author = {Lagos, MJ and Sato, F and Autreto, PAS and Galvao, DS and Rodrigues, V and Ugarte, D},
url = {http://iopscience.iop.org/0957-4484/21/48/485702},
year = {2010},
date = {2010-01-01},
journal = {Nanotechnology},
volume = {21},
number = {48},
pages = {485702},
publisher = {IOP Publishing},
abstract = {We have studied the changes induced by thermal effects in the structural and transport response of Au nanowires generated by mechanical elongation. We have used time-resolved atomic resolution transmission electron microscopy imaging and quantum conductance measurement using a mechanically controllable break junction. Our results showed remarkable differences in the NW evolution for experiments realized at 150 and 300 K, which modifies drastically the conductance response during elongation. Molecular dynamics and electronic transport calculations were used to consistently correlate the observed structural and conductance behavior. These results emphasize that it is essential to take into account the precise atomic arrangement of nanocontacts generated by mechanical stretching to understand electrical transport properties. Also, our study shows that much care must be taken when comparing results obtained in different experimental conditions, mainly different temperatures.
},
keywords = {DFT, Mechanical Properties, Metallic Nanowires, Quantum Transport, TEM},
pubstate = {published},
tppubtype = {article}
}
2008
Coluci, Vitor R; Fonseca, Alexandre F; Galvao, Douglas S; Daraio, Chiara
Entanglement and the nonlinear elastic behavior of forests of coiled carbon nanotubes Journal Article
In: Physical Review Letters, vol. 100, no. 8, pp. 086807, 2008.
Abstract | Links | BibTeX | Tags: Carbon Nanotube Forests, Entanglement, Mechanical Properties, top20
@article{coluci2008entanglement,
title = {Entanglement and the nonlinear elastic behavior of forests of coiled carbon nanotubes},
author = {Coluci, Vitor R and Fonseca, Alexandre F and Galvao, Douglas S and Daraio, Chiara},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.100.086807},
year = {2008},
date = {2008-01-01},
journal = {Physical Review Letters},
volume = {100},
number = {8},
pages = {086807},
publisher = {American Physical Society},
abstract = {Helical or coiled nanostructures have been objects of intense experimental and theoretical studies due to their special electronic and mechanical properties. Recently, it was experimentally reported that the dynamical response of a foamlike forest of coiled carbon nanotubes under mechanical impact exhibits a nonlinear, non-Hertzian behavior, with no trace of plastic deformation. The physical origin of this unusual behavior is not yet fully understood. In this Letter, based on analytical models, we show that the entanglement among neighboring coils in the superior part of the forest surface must be taken into account for a full description of the strongly nonlinear behavior of the impact response of a drop ball onto a forest of coiled carbon nanotubes.},
keywords = {Carbon Nanotube Forests, Entanglement, Mechanical Properties, top20},
pubstate = {published},
tppubtype = {article}
}
2007
Coluci, Vitor R; Pugno, Nicola M; Dantas, Socrates O; Galvao, Douglas S; Jorio, Ado
Atomistic simulations of the mechanical properties of'super'carbon nanotubes Journal Article
In: Nanotechnology, vol. 18, no. 33, pp. 335702, 2007.
Abstract | Links | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, Super Carbons
@article{coluci2007atomistic,
title = {Atomistic simulations of the mechanical properties of'super'carbon nanotubes},
author = {Coluci, Vitor R and Pugno, Nicola M and Dantas, Socrates O and Galvao, Douglas S and Jorio, Ado},
url = {http://iopscience.iop.org/0957-4484/18/33/335702
},
year = {2007},
date = {2007-01-01},
journal = {Nanotechnology},
volume = {18},
number = {33},
pages = {335702},
publisher = {IOP Publishing},
abstract = {The mechanical properties of the so-called 'super' carbon nanotubes (STs) are investigated using classical molecular dynamics simulations. The STs are built from single-walled carbon nanotubes (SWCNTs) connected by Y-like junctions forming an ordered carbon nanotube network that is then rolled into a seamless cylinder. We observed that the ST behaviour under tensile tests is similar to the one presented by fishing nets. This interesting behaviour provides a way to vary the accessible channels to the inner parts of STs by applying an external mechanical load. The Young's modulus is dependent on the ST chirality and it inversely varies with the ST radius. Smaller reduction of breaking strain values due to temperature increase is predicted for zigzag STs compared to SWCNTs. The results show that, for STs with radius ~5 nm, the junctions between the constituent SWCNTs play an important role in the fracture process. The Young's modulus and tensile strength were estimated for hierarchical higher-order STs using scaling laws related to the ST fractal dimension. The obtained mechanical properties suggest that STs may be used in the development of new porous, flexible, and high-strength materials.},
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, Super Carbons},
pubstate = {published},
tppubtype = {article}
}

Fonseca, AD; Malta, CP; Galvao, DS
Elastic Properties of Normal and Binormal Helical Nanowires Proceedings
Warrendale, Pa.; Materials Research Society; 1999, vol. 963, 2007.
Abstract | Links | BibTeX | Tags: Elasticity, Helical Structures, Mechanical Properties, Nanowires
@proceedings{fonseca2007elastic,
title = {Elastic Properties of Normal and Binormal Helical Nanowires},
author = {Fonseca, AD and Malta, CP and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8026852},
year = {2007},
date = {2007-01-01},
journal = {MATERIALS RESEARCH SOCIETY SYMPOSIUM PROCEEDINGS},
volume = {963},
pages = {88},
publisher = {Warrendale, Pa.; Materials Research Society; 1999},
abstract = {A helical nanowire can be defined as being a nanoscopic rod whose axis follows a helical curve in space. In the case of a nanowire with asymmetric cross section, the helical nanostructure can be classified as normal or binormal helix, according to the orientation of the cross section with respect to the helical axis of the structure. In this work, we present a simple model to study the elastic properties of a helical nanowire with asymmetric cross section. We use the framework of the Kirchhoff rod model to obtain an expression relating the Hooke's constant, h, of normal and binormal nanohelices to their geometric features. We also obtain the Young's modulus values. These relations can be used by experimentalists to evaluate the elastic properties of helical nanostructures. We showed that the Hooke's constant of a normal nanohelix is higher than that of a binormal one. We illustrate our results using experimentally obtained nanohelices reported in the literature.},
keywords = {Elasticity, Helical Structures, Mechanical Properties, Nanowires},
pubstate = {published},
tppubtype = {proceedings}
}
Coluci, VR; Dantas, SO; Jorio, A; Galvao, DS
Electronic and Mechanical Properties of Super Carbon Nanotube Networks Proceedings
Warrendale, Pa.; Materials Research Society; 1999, vol. 963, 2007.
Abstract | Links | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, Super Carbons
@proceedings{coluci2007electronic,
title = {Electronic and Mechanical Properties of Super Carbon Nanotube Networks},
author = {Coluci, VR and Dantas, SO and Jorio, A and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8026810&fulltextType=RA&fileId=S1946427400054014},
year = {2007},
date = {2007-01-01},
journal = {MATERIALS RESEARCH SOCIETY SYMPOSIUM PROCEEDINGS},
volume = {963},
pages = {1},
publisher = {Warrendale, Pa.; Materials Research Society; 1999},
abstract = {Eletronic and mechanical properties of ordered carbon nanotube networks are studied using molecular dynamics simulations and tight-binding calculations. These networks are formed by single walled carbon nanotubes (SWNT) regularly connected by junctions. The use of different types of junctions (“Y”-, “X”-like junctions, for example) allows the construction of networks with different symmetries. These networks can be very flexible and the elastic deformation was associated with two main deformation mechanisms (bending and stretching ) of the constituents SWNTs. Rolling up the networks, “super” carbon nanotubes can be constructed. These super-tubes share some of the main electronic features of the SWNT which form them but important changes are predicted (e.g. reduction of bandgap value). Simulations of their deformations under tensile stress have revealed that the super-tubes are softer than the corresponding SWNT and that their rupture occur in higher strain values.},
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, Super Carbons},
pubstate = {published},
tppubtype = {proceedings}
}
Fonseca, Alexandre F; Malta, CP; Galvao, DS
Is it possible to grow amorphous normal nanosprings? Journal Article
In: Nanotechnology, vol. 18, no. 43, pp. 435606, 2007.
Abstract | Links | BibTeX | Tags: Elasticity, Helical Structures, Mechanical Properties, Nanowires
@article{fonseca2007possible,
title = {Is it possible to grow amorphous normal nanosprings?},
author = {Fonseca, Alexandre F and Malta, CP and Galvao, DS},
url = {http://iopscience.iop.org/0957-4484/18/43/435606},
year = {2007},
date = {2007-01-01},
journal = {Nanotechnology},
volume = {18},
number = {43},
pages = {435606},
publisher = {IOP Publishing},
abstract = {Nanosprings have been objects of intense investigations in recent years. They can be classified as normal or binormal depending on the geometry of their cross-section. As normal amorphous nanosprings have not yet been observed experimentally, we have decided to investigate this matter. We discuss the shape of the catalyst in terms of the cross-sectional shape of the nanospring and show that, within the vapor–liquid–solid model, the growth of amorphous binormal nanosprings is energetically favored.},
keywords = {Elasticity, Helical Structures, Mechanical Properties, Nanowires},
pubstate = {published},
tppubtype = {article}
}
Coluci, VR; Dantas, SO; Jorio, A; Galvao, DS
Mechanical properties of carbon nanotube networks by molecular mechanics and impact molecular dynamics calculations Journal Article
In: Physical Review B, vol. 75, no. 7, pp. 075417, 2007.
Abstract | Links | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, Super Carbons
@article{coluci2007mechanical,
title = {Mechanical properties of carbon nanotube networks by molecular mechanics and impact molecular dynamics calculations},
author = {Coluci, VR and Dantas, SO and Jorio, A and Galvao, DS},
url = {http://journals.aps.org/prb/abstract/10.1103/PhysRevB.75.075417},
year = {2007},
date = {2007-01-01},
journal = {Physical Review B},
volume = {75},
number = {7},
pages = {075417},
publisher = {APS},
abstract = {We report a theoretical investigation of the mechanical properties of idealized networks formed by single-walled carbon nanotubes showing crossbar and hexagonal architectures. The study was performed by using molecular mechanics calculations and impact dynamics simulations based on bond-order empirical potential. The studied networks were predicted to have elasticity modulus of ∼10–100GPa and bulk modulus of ∼10GPa. The results show a transition from high to moderate flexibility during the deformation stages. This behavior was associated with the existence of two deformation mechanisms presented by the network related to the nanotube stretching and junction bending processes.},
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, Super Carbons},
pubstate = {published},
tppubtype = {article}
}

Pugno, Nicola; Coluci, V; Galvao, DS
Nanotube-or graphene-based nanoarmors Book Chapter
In: Computational & Experimental Analysis of Damaged Materials, pp. 145-154 , 2007.
Abstract | Links | BibTeX | Tags: Elasticity, Mechanical Properties, Molecular Dynamics, Super Carbons
@inbook{pugno2007nanotube,
title = {Nanotube-or graphene-based nanoarmors},
author = {Pugno, Nicola and Coluci, V and Galvao, DS},
url = {http://www.ing.unitn.it/~pugno/NP_PDF/IV/5-COLUCI07.pdf},
year = {2007},
date = {2007-01-01},
booktitle = {Computational & Experimental Analysis of Damaged Materials},
pages = {145-154 },
abstract = { In this paper, nanoimpacts on hexagonal or
crossbar nanotube networks as well as on graphene
sheets are investigated by elasticity and finite
kinematics or impact molecular dynamic simulations.
A transition from bending to stretching by increasing
the impact kinetic energy of the nanoprojectile is
clearly observed. The analysis suggests that the
investigated nanotextures are ideal for designing
futuristic nanoarmors. },
keywords = {Elasticity, Mechanical Properties, Molecular Dynamics, Super Carbons},
pubstate = {published},
tppubtype = {inbook}
}
crossbar nanotube networks as well as on graphene
sheets are investigated by elasticity and finite
kinematics or impact molecular dynamic simulations.
A transition from bending to stretching by increasing
the impact kinetic energy of the nanoprojectile is
clearly observed. The analysis suggests that the
investigated nanotextures are ideal for designing
futuristic nanoarmors.
Braga, Scheila Furtado; Galvao, Douglas Soares
Molecular dynamics simulation of single wall carbon nanotubes polymerization under compression Journal Article
In: Journal of Computational Chemistry, vol. 28, no. 10, pp. 1724–1734, 2007.
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, Mechanical Properties, Molecular Dynamics, New Structures, Polymerization
@article{braga2007molecular,
title = {Molecular dynamics simulation of single wall carbon nanotubes polymerization under compression},
author = {Braga, Scheila Furtado and Galvao, Douglas Soares},
url = {http://onlinelibrary.wiley.com/store/10.1002/jcc.20684/asset/20684_ftp.pdf?v=1&t=i52l5iyb&s=94cda082eed01cd61890fffe50aad5e26cdda7d1},
year = {2007},
date = {2007-01-01},
journal = {Journal of Computational Chemistry},
volume = {28},
number = {10},
pages = {1724--1734},
publisher = {Wiley Subscription Services, Inc., A Wiley Company},
abstract = {Single wall carbon nanotubes (SWCNTs) often aggregate into bundles of hundreds of weakly interacting
tubes. Their cross-polymerization opens new possibilities for the creation of new super-hard materials. New mechanical
and electronic properties are expected from these condensed structures, as well as novel potential applications. Previous
theoretical results presented geometric modifications involving changes in the radial section of the compressed tubes
as the explanation to the experimental measurements of structural changes during tube compression. We report here
results from molecular dynamics simulations of the SWCNTs polymerization for small diameter arm chair tubes under
compression. Hydrostatic and piston-type compression of SWCNTs have been simulated for different temperatures and
rates of compression. Our results indicate that large diameter tubes (10,10) are unlike to polymerize while small diameter
ones (around 5 Å) polymerize even at room temperature. Other interesting results are the observation of the appearance
of spontaneous scroll-like structures and also the so-called tubulane motifs, which were predicted in the literature more
than a decade ago},
keywords = {Carbon Nanotubes, Mechanical Properties, Molecular Dynamics, New Structures, Polymerization},
pubstate = {published},
tppubtype = {article}
}
tubes. Their cross-polymerization opens new possibilities for the creation of new super-hard materials. New mechanical
and electronic properties are expected from these condensed structures, as well as novel potential applications. Previous
theoretical results presented geometric modifications involving changes in the radial section of the compressed tubes
as the explanation to the experimental measurements of structural changes during tube compression. We report here
results from molecular dynamics simulations of the SWCNTs polymerization for small diameter arm chair tubes under
compression. Hydrostatic and piston-type compression of SWCNTs have been simulated for different temperatures and
rates of compression. Our results indicate that large diameter tubes (10,10) are unlike to polymerize while small diameter
ones (around 5 Å) polymerize even at room temperature. Other interesting results are the observation of the appearance
of spontaneous scroll-like structures and also the so-called tubulane motifs, which were predicted in the literature more
than a decade ago
2006
Coluci, Vitor R; Dantas, Socrates O; Jorio, Ado; Galvao, Douglas S.
Electronic and Mechanical Properties of Super Carbon Nanotube Networks Proceedings
Cambridge University Press, vol. 963, 2006.
Abstract | Links | BibTeX | Tags: Mechanical Properties, Molecular Dynamics, Super Carbons
@proceedings{coluci2006electronic,
title = {Electronic and Mechanical Properties of Super Carbon Nanotube Networks},
author = {Coluci, Vitor R and Dantas, Socrates O and Jorio, Ado and Galvao, Douglas S.},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8026810&fileId=S1946427400054014},
year = {2006},
date = {2006-01-01},
journal = {MRS Proceedings},
volume = {963},
pages = {0963--Q15},
publisher = {Cambridge University Press},
abstract = {Eletronic and mechanical properties of ordered carbon nanotube networks are studied using molecular dynamics simulations and tight-binding calculations. These networks are formed by single walled carbon nanotubes (SWNT) regularly connected by junctions. The use of different types of junctions (“Y”-, “X”-like junctions, for example) allows the construction of networks with different symmetries. These networks can be very flexible and the elastic deformation was associated with two main deformation mechanisms (bending and stretching ) of the constituents SWNTs. Rolling up the networks, “super” carbon nanotubes can be constructed. These super-tubes share some of the main electronic features of the SWNT which form them but important changes are predicted (e.g. reduction of bandgap value). Simulations of their deformations under tensile stress have revealed that the super-tubes are softer than the corresponding SWNT and that their rupture occur in higher strain values.},
keywords = {Mechanical Properties, Molecular Dynamics, Super Carbons},
pubstate = {published},
tppubtype = {proceedings}
}
Coluci, Vitor R; Galvao, Douglas S; Jorio, A
Geometric and electronic structure of carbon nanotube networks:'super'-carbon nanotubes Journal Article
In: Nanotechnology, vol. 17, no. 3, pp. 617, 2006.
Abstract | Links | BibTeX | Tags: DFT, Mechanical Properties, Molecular Dynamics, Super Carbons
@article{coluci2006geometric,
title = {Geometric and electronic structure of carbon nanotube networks:'super'-carbon nanotubes},
author = {Coluci, Vitor R and Galvao, Douglas S and Jorio, A},
url = {http://iopscience.iop.org/0957-4484/17/3/001},
year = {2006},
date = {2006-01-01},
journal = {Nanotechnology},
volume = {17},
number = {3},
pages = {617},
publisher = {IoP Publishing},
abstract = {Structures of the so-called super-carbon nanotubes are proposed. These structures are built from single walled carbon nanotubes connected by Y-like junctions forming a 'super'-sheet that is then rolled into a seamless cylinder. Such a procedure can be repeated several times, generating a fractal structure. This procedure is not limited to carbon nanotubes, and can be easily modified for application to other systems. Tight binding total energy and density of states calculations showed that the 'super'-sheets and tubes are stable and predicted to present metallic and semiconducting behaviour.},
keywords = {DFT, Mechanical Properties, Molecular Dynamics, Super Carbons},
pubstate = {published},
tppubtype = {article}
}
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ

