
de Sousa, JM; Aguiar, AL; Girao, EC; Fonseca, Alexandre F; AG Filho, Souza; Galvao, Douglas S
Mechanical Properties and Fracture Patterns of Pentagraphene Membranes (under review) Journal Article
In: 2019.
@article{deSousa2019,
title = {Mechanical Properties and Fracture Patterns of Pentagraphene Membranes (under review)},
author = {de Sousa, JM and Aguiar, AL and Girao, EC and Fonseca, Alexandre F and AG Filho, Souza and Galvao, Douglas S},
year = {2019},
date = {2019-01-05},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
de Sousa, J. M.; Aguiar, A. L.; Girao, E. C.; Fonseca, Alexandre F.; Filho, A. G. Souza; Galvao, Douglas S.
Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 97-102, 2018.
@article{deSousa2018b,
title = {Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study},
author = {J. M. de Sousa and A. L. Aguiar and E. C. Girao and Alexandre F. Fonseca and A. G. Souza Filho and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-pentagraphenebased-nanotubes-a-molecular-dynamics-study/289AB70DADF20059BB8FCC9EF07B97AB},
doi = { https://doi.org/10.1557/adv.2018.160},
year = {2018},
date = {2018-02-06},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = {97-102},
abstract = {The study of the mechanical properties of nanostructured systems has gained importance in theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of the strongest nanomaterials found in nature, with Young’s Modulus (EY) in the order 1.25 TPa. One interesting question is about the possibility of generating new nanostructures with 1D symmetry and with similar and/or superior CNT properties. In this work, we present a study on the dynamical, structural, mechanical properties, fracture patterns and EY values for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized states) in the same form that CNTs are formed from rolling up graphene membranes. We carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We have considered zigzag-like and armchair-like PGNTs of different diameters. Our results show that PGNTs present EY ∼ 800 GPa with distinct elastic behavior in relation to CNTs, mainly associated with mechanical failure, chirality dependent fracture patterns and extensive structural reconstructions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Azevedo, David L.; Bizao, Rafael A.; Galvao, Douglas S.
Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets Online
2018, (preprint arXiv:1801.05346).
@online{Azevedo2018b,
title = {Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets},
author = {David L. Azevedo and Rafael A. Bizao and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.05346},
year = {2018},
date = {2018-01-18},
abstract = {The superior mechanical properties and low density of carbon nanostructures make them promising ballistic protection materials, stimulating investigations on their high-strain-rate behavior. Recent experiments and simulations revealed graphene possesses exceptional energy absorption properties. In this work, we analyzed through fully atomistic molecular dynamics simulations the ballistic performance of a carbon-based material recently proposed named penta-graphene. Our results show that the fracture pattern is more spherical (no petals formation like observed for graphene). The estimated penetration energy for pentagraphene structures considered here was of 37.69 MJ/Kg, far superior to graphene (29.8 MJ/Kg) under same conditions. These preliminary results are suggestive that pentagraphene could be an excellent material for ballistic applications.},
note = {preprint arXiv:1801.05346},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
de Sousa, J. M.; Aguiar, A. L.; Girao, E. C.; Fonseca, Alexandre F.; Filho, A. G. Sousa; Galvao, Douglas S.
Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study Online
2018, (preprint arXiv:1801.04269).
@online{deSousa2018d,
title = {Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study},
author = {J. M. de Sousa and A. L. Aguiar and E. C. Girao and Alexandre F. Fonseca and A. G. Sousa Filho and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.04269},
year = {2018},
date = {2018-01-15},
abstract = {The study of the mechanical properties of nanostructured systems has gained importance in
theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of
the strongest nanomaterials found in nature, with Young's Modulus (YM) in the order 1.25
TPa. One interesting question is about the possibility of generating new nanostructures with
1D symmetry and with similar and/or superior CNT properties. In this work, we present a
study on the dynamical, structural, mechanical properties, fracture patterns and YM values
for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These
tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional
structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized
states) in the same form that CNTs are formed from rolling up graphene membranes. We
carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We
have considered zigzag-like and armchair-like PGNTs of different diameters. Our results
show that PGNTs present YM ~ 800 GPa with distinct elastic behavior in relation to CNTs,
mainly associated with mechanical failure, chirality dependent fracture patterns and extensive
structural reconstructions},
note = {preprint arXiv:1801.04269},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of
the strongest nanomaterials found in nature, with Young's Modulus (YM) in the order 1.25
TPa. One interesting question is about the possibility of generating new nanostructures with
1D symmetry and with similar and/or superior CNT properties. In this work, we present a
study on the dynamical, structural, mechanical properties, fracture patterns and YM values
for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These
tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional
structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized
states) in the same form that CNTs are formed from rolling up graphene membranes. We
carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We
have considered zigzag-like and armchair-like PGNTs of different diameters. Our results
show that PGNTs present YM ~ 800 GPa with distinct elastic behavior in relation to CNTs,
mainly associated with mechanical failure, chirality dependent fracture patterns and extensive
structural reconstructions
de Sousa, Jose M.; Aguiar, Acrisio L.; Girao, Eduardo C.; Fonseca, Alexandre F.; Antonio G. Souza Filho,; Galvao, Douglas S.
Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study Online
2018, (preprint arXiv:1801.04269).
@online{deSousa2018f,
title = {Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study},
author = {Jose M. de Sousa and Acrisio L. Aguiar and Eduardo C. Girao and Alexandre F. Fonseca and Antonio G. Souza Filho, and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.04269},
year = {2018},
date = {2018-01-12},
abstract = {The study of the mechanical properties of nanostructured systems has gained importance in theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of the strongest nanomaterials found in nature, with Young's Modulus (YM) in the order 1.25 TPa. One interesting question is about the possibility of generating new nanostructures with 1D symmetry and with similar and/or superior CNT properties. In this work, we present a study on the dynamical, structural, mechanical properties, fracture patterns and YM values for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized states) in the same form that CNTs are formed from rolling up graphene membranes. We carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We have considered zigzag-like and armchair-like PGNTs of different diameters. Our results show that PGNTs present YM ~ 800 GPa with distinct elastic behavior in relation to CNTs, mainly associated with mechanical failure, chirality dependent fracture patterns and extensive structural reconstructions.},
note = {preprint arXiv:1801.04269},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Azevedo, David L.; Bizao, Rafael A.; Galvao, Douglas S.
Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets Journal Article
In: MRS Advances, vol. 3, no. 8-9, pp. 431-435, 2018.
@article{Azevedo2018,
title = {Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets},
author = {David L. Azevedo and Rafael A. Bizao and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/molecular-dynamics-simulations-of-ballistic-penetration-of-pentagraphene-sheets/8759C0815840EDE83896EF4A17278228},
doi = {https://doi.org/10.1557/adv.2018.61},
year = {2018},
date = {2018-01-06},
journal = {MRS Advances},
volume = {3},
number = {8-9},
pages = {431-435},
abstract = {The search for new materials with low density and superior mechanical properties is a very intense and stimulating investigation area. These new materials could provide potential application for ballistic protection. Recent experiments and simulations revealed graphene possesses exceptional energy absorption properties. In this work, we analysed through fully atomistic molecular dynamics simulations the ballistic performance of a carbon-based material recently proposed named penta-graphene. Our results show that the fracture pattern is more spherical (no petals formation like observed for graphene). The estimated penetration energy for single-layer penta-graphene structures obtained here was d_1penta∼37.7 MJ/kg, and is comparable with recently results obtained for graphene: d_(1graphene)∼29.0 MJ/kg and d_(1graphene)∼40.8 MJ/kg under similar conditions. These preliminary results are suggestive that penta-graphene could be an excellent material for ballistic applications.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
de Sousa, JM; Aguiar, AL; Girao, EC; Fonseca, Alexandre F; AG Filho, Souza; Galvao, Douglas S
Mechanical Properties and Fracture Patterns of Pentagraphene Membranes Online
2017, (preprint arXiv:1703.03789).
@online{deSousa2017,
title = {Mechanical Properties and Fracture Patterns of Pentagraphene Membranes},
author = {de Sousa, JM and Aguiar, AL and Girao, EC and Fonseca, Alexandre F and AG Filho, Souza and Galvao, Douglas S},
url = {https://arxiv.org/abs/1703.03789},
year = {2017},
date = {2017-03-10},
abstract = {Recently, a new two-dimensional carbon allotrope called pentagraphene (PG) was
proposed. PG exhibits mechanical and electronic interesting properties, including typical
band gap values of semiconducting materials. PG has a Cairo-tiling-like 2D lattice
of non coplanar pentagons and its mechanical properties have not been yet fully investigated.
In this work, we combined density functional theory (DFT) calculations and
reactive molecular dynamics (MD) simulations to investigate the mechanical properties
and fracture patterns of PG membranes under tensile strain. We show that PG
membranes can hold up to 20% of strain and that fracture occurs only after substantial
dynamical bond breaking and the formation of 7, 8 and 11 carbon rings and carbon
chains. The stress-strain behavior was observed to follow two regimes, one exhibiting linear elasticity followed by a plastic one, involving carbon atom re-hybridization with
the formation of carbon rings and chains. Our results also show that mechanically
induced structural transitions from PG to graphene is unlikely to occur, in contrast to
what was previously speculated in the literature.},
note = {preprint arXiv:1703.03789},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
proposed. PG exhibits mechanical and electronic interesting properties, including typical
band gap values of semiconducting materials. PG has a Cairo-tiling-like 2D lattice
of non coplanar pentagons and its mechanical properties have not been yet fully investigated.
In this work, we combined density functional theory (DFT) calculations and
reactive molecular dynamics (MD) simulations to investigate the mechanical properties
and fracture patterns of PG membranes under tensile strain. We show that PG
membranes can hold up to 20% of strain and that fracture occurs only after substantial
dynamical bond breaking and the formation of 7, 8 and 11 carbon rings and carbon
chains. The stress-strain behavior was observed to follow two regimes, one exhibiting linear elasticity followed by a plastic one, involving carbon atom re-hybridization with
the formation of carbon rings and chains. Our results also show that mechanically
induced structural transitions from PG to graphene is unlikely to occur, in contrast to
what was previously speculated in the literature.
2019
de Sousa, JM; Aguiar, AL; Girao, EC; Fonseca, Alexandre F; AG Filho, Souza; Galvao, Douglas S
Mechanical Properties and Fracture Patterns of Pentagraphene Membranes (under review) Journal Article
In: 2019.
BibTeX | Tags: Fracture, Molecular Dynamics, pentagraphene
@article{deSousa2019,
title = {Mechanical Properties and Fracture Patterns of Pentagraphene Membranes (under review)},
author = {de Sousa, JM and Aguiar, AL and Girao, EC and Fonseca, Alexandre F and AG Filho, Souza and Galvao, Douglas S},
year = {2019},
date = {2019-01-05},
keywords = {Fracture, Molecular Dynamics, pentagraphene},
pubstate = {published},
tppubtype = {article}
}
2018
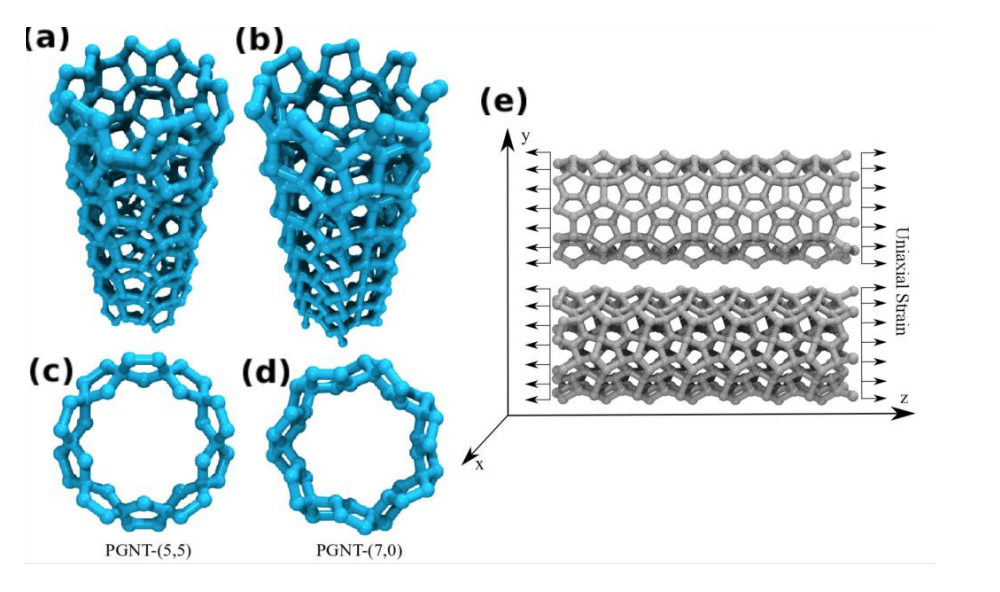
de Sousa, J. M.; Aguiar, A. L.; Girao, E. C.; Fonseca, Alexandre F.; Filho, A. G. Souza; Galvao, Douglas S.
Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 97-102, 2018.
Abstract | Links | BibTeX | Tags: Fracture, Molecular Dynamics, pentagraphene
@article{deSousa2018b,
title = {Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study},
author = {J. M. de Sousa and A. L. Aguiar and E. C. Girao and Alexandre F. Fonseca and A. G. Souza Filho and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-pentagraphenebased-nanotubes-a-molecular-dynamics-study/289AB70DADF20059BB8FCC9EF07B97AB},
doi = { https://doi.org/10.1557/adv.2018.160},
year = {2018},
date = {2018-02-06},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = {97-102},
abstract = {The study of the mechanical properties of nanostructured systems has gained importance in theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of the strongest nanomaterials found in nature, with Young’s Modulus (EY) in the order 1.25 TPa. One interesting question is about the possibility of generating new nanostructures with 1D symmetry and with similar and/or superior CNT properties. In this work, we present a study on the dynamical, structural, mechanical properties, fracture patterns and EY values for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized states) in the same form that CNTs are formed from rolling up graphene membranes. We carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We have considered zigzag-like and armchair-like PGNTs of different diameters. Our results show that PGNTs present EY ∼ 800 GPa with distinct elastic behavior in relation to CNTs, mainly associated with mechanical failure, chirality dependent fracture patterns and extensive structural reconstructions.},
keywords = {Fracture, Molecular Dynamics, pentagraphene},
pubstate = {published},
tppubtype = {article}
}
Azevedo, David L.; Bizao, Rafael A.; Galvao, Douglas S.
Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets Online
2018, (preprint arXiv:1801.05346).
Abstract | Links | BibTeX | Tags: Fracture, Molecular Dynamics, pentagraphene
@online{Azevedo2018b,
title = {Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets},
author = {David L. Azevedo and Rafael A. Bizao and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.05346},
year = {2018},
date = {2018-01-18},
abstract = {The superior mechanical properties and low density of carbon nanostructures make them promising ballistic protection materials, stimulating investigations on their high-strain-rate behavior. Recent experiments and simulations revealed graphene possesses exceptional energy absorption properties. In this work, we analyzed through fully atomistic molecular dynamics simulations the ballistic performance of a carbon-based material recently proposed named penta-graphene. Our results show that the fracture pattern is more spherical (no petals formation like observed for graphene). The estimated penetration energy for pentagraphene structures considered here was of 37.69 MJ/Kg, far superior to graphene (29.8 MJ/Kg) under same conditions. These preliminary results are suggestive that pentagraphene could be an excellent material for ballistic applications.},
note = {preprint arXiv:1801.05346},
keywords = {Fracture, Molecular Dynamics, pentagraphene},
pubstate = {published},
tppubtype = {online}
}
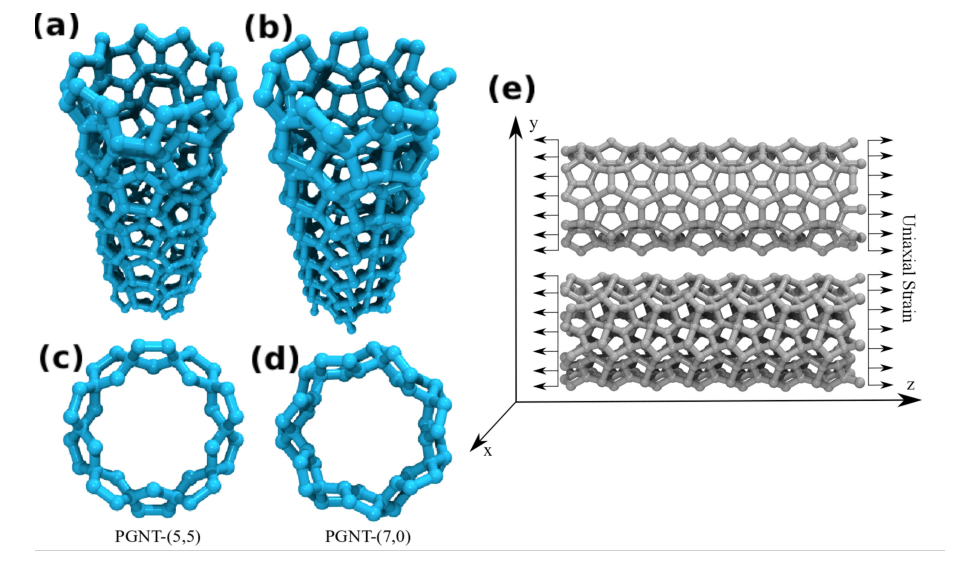
de Sousa, J. M.; Aguiar, A. L.; Girao, E. C.; Fonseca, Alexandre F.; Filho, A. G. Sousa; Galvao, Douglas S.
Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study Online
2018, (preprint arXiv:1801.04269).
Abstract | Links | BibTeX | Tags: Fracture, Molecular Dynamics, Nanotubes, pentagraphene
@online{deSousa2018d,
title = {Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study},
author = {J. M. de Sousa and A. L. Aguiar and E. C. Girao and Alexandre F. Fonseca and A. G. Sousa Filho and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.04269},
year = {2018},
date = {2018-01-15},
abstract = {The study of the mechanical properties of nanostructured systems has gained importance in
theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of
the strongest nanomaterials found in nature, with Young's Modulus (YM) in the order 1.25
TPa. One interesting question is about the possibility of generating new nanostructures with
1D symmetry and with similar and/or superior CNT properties. In this work, we present a
study on the dynamical, structural, mechanical properties, fracture patterns and YM values
for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These
tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional
structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized
states) in the same form that CNTs are formed from rolling up graphene membranes. We
carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We
have considered zigzag-like and armchair-like PGNTs of different diameters. Our results
show that PGNTs present YM ~ 800 GPa with distinct elastic behavior in relation to CNTs,
mainly associated with mechanical failure, chirality dependent fracture patterns and extensive
structural reconstructions},
note = {preprint arXiv:1801.04269},
keywords = {Fracture, Molecular Dynamics, Nanotubes, pentagraphene},
pubstate = {published},
tppubtype = {online}
}
theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of
the strongest nanomaterials found in nature, with Young's Modulus (YM) in the order 1.25
TPa. One interesting question is about the possibility of generating new nanostructures with
1D symmetry and with similar and/or superior CNT properties. In this work, we present a
study on the dynamical, structural, mechanical properties, fracture patterns and YM values
for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These
tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional
structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized
states) in the same form that CNTs are formed from rolling up graphene membranes. We
carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We
have considered zigzag-like and armchair-like PGNTs of different diameters. Our results
show that PGNTs present YM ~ 800 GPa with distinct elastic behavior in relation to CNTs,
mainly associated with mechanical failure, chirality dependent fracture patterns and extensive
structural reconstructions
de Sousa, Jose M.; Aguiar, Acrisio L.; Girao, Eduardo C.; Fonseca, Alexandre F.; Antonio G. Souza Filho,; Galvao, Douglas S.
Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study Online
2018, (preprint arXiv:1801.04269).
Abstract | Links | BibTeX | Tags: Fracture, Molecular Dynamics, pentagraphene
@online{deSousa2018f,
title = {Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study},
author = {Jose M. de Sousa and Acrisio L. Aguiar and Eduardo C. Girao and Alexandre F. Fonseca and Antonio G. Souza Filho, and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.04269},
year = {2018},
date = {2018-01-12},
abstract = {The study of the mechanical properties of nanostructured systems has gained importance in theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of the strongest nanomaterials found in nature, with Young's Modulus (YM) in the order 1.25 TPa. One interesting question is about the possibility of generating new nanostructures with 1D symmetry and with similar and/or superior CNT properties. In this work, we present a study on the dynamical, structural, mechanical properties, fracture patterns and YM values for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized states) in the same form that CNTs are formed from rolling up graphene membranes. We carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We have considered zigzag-like and armchair-like PGNTs of different diameters. Our results show that PGNTs present YM ~ 800 GPa with distinct elastic behavior in relation to CNTs, mainly associated with mechanical failure, chirality dependent fracture patterns and extensive structural reconstructions.},
note = {preprint arXiv:1801.04269},
keywords = {Fracture, Molecular Dynamics, pentagraphene},
pubstate = {published},
tppubtype = {online}
}

Azevedo, David L.; Bizao, Rafael A.; Galvao, Douglas S.
Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets Journal Article
In: MRS Advances, vol. 3, no. 8-9, pp. 431-435, 2018.
Abstract | Links | BibTeX | Tags: Fracture, Molecular Dynamics, pentagraphene
@article{Azevedo2018,
title = {Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets},
author = {David L. Azevedo and Rafael A. Bizao and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/molecular-dynamics-simulations-of-ballistic-penetration-of-pentagraphene-sheets/8759C0815840EDE83896EF4A17278228},
doi = {https://doi.org/10.1557/adv.2018.61},
year = {2018},
date = {2018-01-06},
journal = {MRS Advances},
volume = {3},
number = {8-9},
pages = {431-435},
abstract = {The search for new materials with low density and superior mechanical properties is a very intense and stimulating investigation area. These new materials could provide potential application for ballistic protection. Recent experiments and simulations revealed graphene possesses exceptional energy absorption properties. In this work, we analysed through fully atomistic molecular dynamics simulations the ballistic performance of a carbon-based material recently proposed named penta-graphene. Our results show that the fracture pattern is more spherical (no petals formation like observed for graphene). The estimated penetration energy for single-layer penta-graphene structures obtained here was d_1penta∼37.7 MJ/kg, and is comparable with recently results obtained for graphene: d_(1graphene)∼29.0 MJ/kg and d_(1graphene)∼40.8 MJ/kg under similar conditions. These preliminary results are suggestive that penta-graphene could be an excellent material for ballistic applications.},
keywords = {Fracture, Molecular Dynamics, pentagraphene},
pubstate = {published},
tppubtype = {article}
}
2017
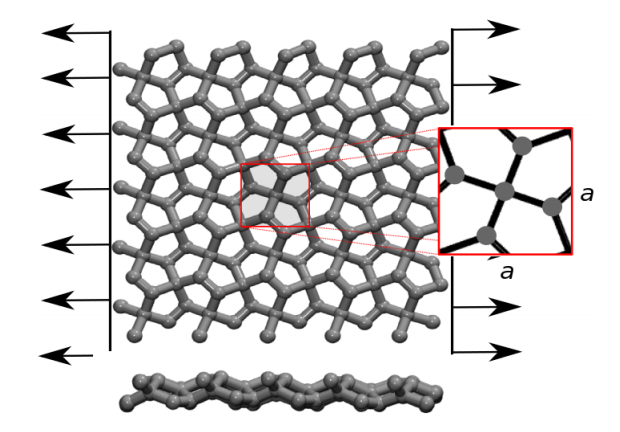
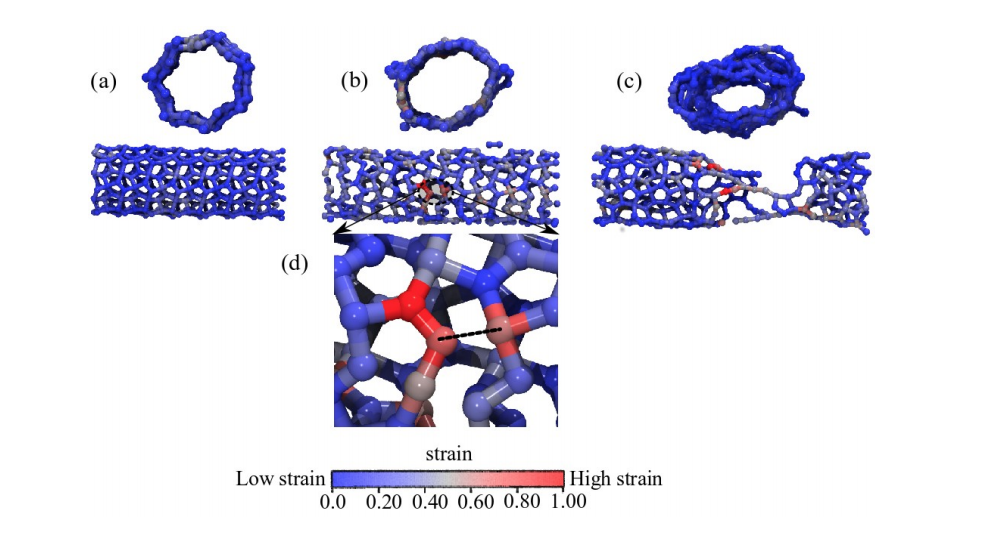
de Sousa, JM; Aguiar, AL; Girao, EC; Fonseca, Alexandre F; AG Filho, Souza; Galvao, Douglas S
Mechanical Properties and Fracture Patterns of Pentagraphene Membranes Online
2017, (preprint arXiv:1703.03789).
Abstract | Links | BibTeX | Tags: DFT, Mechanical Properties, Molecular Dynamics, pentagraphene
@online{deSousa2017,
title = {Mechanical Properties and Fracture Patterns of Pentagraphene Membranes},
author = {de Sousa, JM and Aguiar, AL and Girao, EC and Fonseca, Alexandre F and AG Filho, Souza and Galvao, Douglas S},
url = {https://arxiv.org/abs/1703.03789},
year = {2017},
date = {2017-03-10},
abstract = {Recently, a new two-dimensional carbon allotrope called pentagraphene (PG) was
proposed. PG exhibits mechanical and electronic interesting properties, including typical
band gap values of semiconducting materials. PG has a Cairo-tiling-like 2D lattice
of non coplanar pentagons and its mechanical properties have not been yet fully investigated.
In this work, we combined density functional theory (DFT) calculations and
reactive molecular dynamics (MD) simulations to investigate the mechanical properties
and fracture patterns of PG membranes under tensile strain. We show that PG
membranes can hold up to 20% of strain and that fracture occurs only after substantial
dynamical bond breaking and the formation of 7, 8 and 11 carbon rings and carbon
chains. The stress-strain behavior was observed to follow two regimes, one exhibiting linear elasticity followed by a plastic one, involving carbon atom re-hybridization with
the formation of carbon rings and chains. Our results also show that mechanically
induced structural transitions from PG to graphene is unlikely to occur, in contrast to
what was previously speculated in the literature.},
note = {preprint arXiv:1703.03789},
keywords = {DFT, Mechanical Properties, Molecular Dynamics, pentagraphene},
pubstate = {published},
tppubtype = {online}
}
proposed. PG exhibits mechanical and electronic interesting properties, including typical
band gap values of semiconducting materials. PG has a Cairo-tiling-like 2D lattice
of non coplanar pentagons and its mechanical properties have not been yet fully investigated.
In this work, we combined density functional theory (DFT) calculations and
reactive molecular dynamics (MD) simulations to investigate the mechanical properties
and fracture patterns of PG membranes under tensile strain. We show that PG
membranes can hold up to 20% of strain and that fracture occurs only after substantial
dynamical bond breaking and the formation of 7, 8 and 11 carbon rings and carbon
chains. The stress-strain behavior was observed to follow two regimes, one exhibiting linear elasticity followed by a plastic one, involving carbon atom re-hybridization with
the formation of carbon rings and chains. Our results also show that mechanically
induced structural transitions from PG to graphene is unlikely to occur, in contrast to
what was previously speculated in the literature.
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ

