
JM; Sousa, Bizao
Elastic Properties of Graphyne-based Nanotubes Online
2019, (ArXiv preprint.).
@online{deSousa2019b,
title = {Elastic Properties of Graphyne-based Nanotubes},
author = {de Sousa, JM; , Bizao, RA; Sousa Filho, VP; Aguiar, AL; Coluci, VR; Pugno, NM; Girao, EC; Souza Filho, AG; Galvao, DS},
url = {https://arxiv.org/pdf/1905.02104.pdf},
year = {2019},
date = {2019-04-07},
abstract = {Graphyne nanotubes (GNTs) are nanostructures obtained from rolled up graphyne sheets,
in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes
are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to
conventional CNTs, GNTs can present different chiralities and electronic properties. Because
of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their
mechanical properties. In this work, we studied the mechanical response of GNTs under
tensile stress using fully atomistic molecular dynamics simulations and density functional
theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs
at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller
ultimate strength and Young’s modulus values. This is a consequence of the combined
effects of the existence of triple bonds and increased porosity/flexibility due to the presence
of acetylenic groups.},
note = {ArXiv preprint.},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes
are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to
conventional CNTs, GNTs can present different chiralities and electronic properties. Because
of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their
mechanical properties. In this work, we studied the mechanical response of GNTs under
tensile stress using fully atomistic molecular dynamics simulations and density functional
theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs
at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller
ultimate strength and Young’s modulus values. This is a consequence of the combined
effects of the existence of triple bonds and increased porosity/flexibility due to the presence
of acetylenic groups.
JM; Sousa, Bizao
Elastic Properties of Graphyne-Based Nanotubes Journal Article
In: Computational Materials Science, vol. 170, pp. 109153, 2019.
@article{deSousa2019c,
title = {Elastic Properties of Graphyne-Based Nanotubes},
author = {de Sousa, JM; , Bizao, RA; Sousa Filho, VP; Aguiar, AL; Coluci, VR; Pugno, NM; Girao, EC; Souza Filho, AG; Galvao, DS},
url = {https://www.sciencedirect.com/science/article/pii/S0927025619304525?dgcid=coauthor#s0040},
doi = {10.1016/j.commatsci.2019.109153},
year = {2019},
date = {2019-04-03},
journal = {Computational Materials Science},
volume = {170},
pages = {109153},
abstract = {Graphyne nanotubes (GNTs) are nanostructures obtained from rolled up graphyne sheets, in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to conventional CNTs, GNTs can present different chiralities and electronic properties. Because of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their mechanical properties. In this work, we studied the mechanical response of GNTs under tensile stress using fully atomistic molecular dynamics simulations and density functional theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller ultimate strength and Young’s modulus values. This is a consequence of the combined effects of the existence of triple bonds and increased porosity/flexibility due to the presence of acetylenic groups.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
de Sousa, J. M.; Aguiar, A. L.; Girao, E. C.; Fonseca, Alexandre F.; Filho, A. G. Sousa; Galvao, Douglas S.
Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study Online
2018, (preprint arXiv:1801.04269).
@online{deSousa2018d,
title = {Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study},
author = {J. M. de Sousa and A. L. Aguiar and E. C. Girao and Alexandre F. Fonseca and A. G. Sousa Filho and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.04269},
year = {2018},
date = {2018-01-15},
abstract = {The study of the mechanical properties of nanostructured systems has gained importance in
theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of
the strongest nanomaterials found in nature, with Young's Modulus (YM) in the order 1.25
TPa. One interesting question is about the possibility of generating new nanostructures with
1D symmetry and with similar and/or superior CNT properties. In this work, we present a
study on the dynamical, structural, mechanical properties, fracture patterns and YM values
for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These
tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional
structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized
states) in the same form that CNTs are formed from rolling up graphene membranes. We
carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We
have considered zigzag-like and armchair-like PGNTs of different diameters. Our results
show that PGNTs present YM ~ 800 GPa with distinct elastic behavior in relation to CNTs,
mainly associated with mechanical failure, chirality dependent fracture patterns and extensive
structural reconstructions},
note = {preprint arXiv:1801.04269},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of
the strongest nanomaterials found in nature, with Young's Modulus (YM) in the order 1.25
TPa. One interesting question is about the possibility of generating new nanostructures with
1D symmetry and with similar and/or superior CNT properties. In this work, we present a
study on the dynamical, structural, mechanical properties, fracture patterns and YM values
for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These
tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional
structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized
states) in the same form that CNTs are formed from rolling up graphene membranes. We
carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We
have considered zigzag-like and armchair-like PGNTs of different diameters. Our results
show that PGNTs present YM ~ 800 GPa with distinct elastic behavior in relation to CNTs,
mainly associated with mechanical failure, chirality dependent fracture patterns and extensive
structural reconstructions
Amelia HC Hart Ryota Koizumi, Gustavo Brunetto
Mechano-chemical stabilization of three-dimensional carbon nanotube aggregates Journal Article
In: Carbon, vol. 110, pp. 27-33, 2016.
@article{koizumi2016mechano,
title = {Mechano-chemical stabilization of three-dimensional carbon nanotube aggregates},
author = {Ryota Koizumi, Amelia HC Hart, Gustavo Brunetto, Sanjit Bhowmick, Peter S Owuor, John T Hamel, Anieph X Gentles, Sehmus Ozden, Jun Lou, Robert Vajtai, SA Syed Asif, Douglas S Galvão, CS Tiwary, PM Ajayan},
url = {www.sciencedirect.com/science/article/pii/S0008622316307400},
doi = {10.1016/j.carbon.2016.08.085},
year = {2016},
date = {2016-08-21},
journal = {Carbon},
volume = {110},
pages = {27-33},
abstract = {Here we report a combined study of experiments and simulations to understand how chemical functional groups can mechanically stabilize aggregates of carbon nanotubes (CNTs). Ultralow density aggregates of chemically functionalized CNTs, in the form of macro-scale spheres made by freeze-drying method, show mechanical stabilization and near complete elastic recovery during deformation. Simulations of interacting functionalized carbon nanotube aggregates show better structural retention compared to non-functionalized CNTs under compression, suggesting that the atomic-level interactions between functional groups on adjoining CNTs help maintain structural rigidity and elastic response during loading. Aggregates of non-functionalized CNTs collapses under similar loading conditions. The dynamic mechanical responses of CNT macrostructures and mechano-chemical stabilization are directly observed using in-situ deformation inside a scanning electron microscope.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
G. Brunetto J.M. de Sousa, V. R. Coluci
Torsional “superplasticity” of graphyne nanotubes Journal Article
In: Carbon, vol. 96, pp. 14-19, 2016.
@article{deSousa2016,
title = {Torsional “superplasticity” of graphyne nanotubes},
author = {J.M. de Sousa, G. Brunetto, V.R. Coluci, D.S. Galvao },
url = {http://www.sciencedirect.com/science/article/pii/S000862231530258X},
doi = { http://dx.doi.org/10.1016/j.carbon.2015.09.039},
year = {2016},
date = {2016-01-01},
journal = {Carbon},
volume = {96},
pages = {14-19},
abstract = {Graphyne is a planar two-dimensional carbon allotrope formed by atoms in sp, sp2, and sp3 hybridized states. Topologically graphyne nanotubes (GNTs) can be considered as cylindrically rolled up graphyne sheets, similarly as carbon nanotubes (CNTs) can be considered rolled up graphene sheets. Due to the presence of single, double, and triple bonds, GNTs exhibit porous sidewalls that can be exploited in many diverse applications. In this work, we investigated the mechanical behavior of GNTs under torsional strains through reactive molecular dynamics simulations. Our results show that GNTs are more flexible than CNTs and exhibit “superplasticit”, with fracture angles that are up to 35 times higher than the ones reported to CNTs. This GNT “superplastic” behavior can be explained in terms of irreversible recon- struction processes (mainly associated with the triple bonds) that occur during torsional strains.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Gustavo Brunetto Jose M. de Sousa, Vitor R. Coluci
Torsional "Superplasticity" of Graphyne Nanotubes Online
2015, (ArXiv reprint of Torsional "Superplasticity" of Graphyne Nanotubes, published in Carbon 96, 14 (2016).).
@online{deSousa2015b,
title = {Torsional "Superplasticity" of Graphyne Nanotubes},
author = {Jose M. de Sousa, Gustavo Brunetto, Vitor R. Coluci, Douglas S. Galvao},
url = {http://arxiv.org/abs/1509.08746},
year = {2015},
date = {2015-09-29},
abstract = {Graphyne is a planar two-dimensional carbon allotrope formed by atoms in sp, sp2, and sp3 hybridized states. Topologically graphyne nanotubes (GNTs) can be considered as cylindrically rolled up graphyne sheets, similarly as carbon nanotubes (CNTs) can be considered rolled up graphene sheets. Due to the presence of single, double, and triple bonds, GNTs exhibit porous sidewalls that can be exploited in many diverse applications. In this work, we investigated the mechanical behavior of GNTs under torsional strains through reactive molecular dynamics simulations. Our results show that GNTs are more flexible than CNTs and exhibit 'superplasticity', with fracture angles that are up to 35 times higher than the ones reported to CNTs. This GNT 'superplastic' behavior can be explained in terms of irreversible reconstruction processes (mainly associated with the triple bonds) that occur during torsional strains.},
note = {ArXiv reprint of Torsional "Superplasticity" of Graphyne Nanotubes, published in Carbon 96, 14 (2016).},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Perim, E; Paupitz, R; Botari, T; Galvao, DS
One-dimensional silicon and germanium nanostructures with no carbon analogues Journal Article
In: Physical Chemistry Chemical Physics, vol. 16, no. 44, pp. 24570–24574, 2014.
@article{perim2014one,
title = {One-dimensional silicon and germanium nanostructures with no carbon analogues},
author = {Perim, E and Paupitz, R and Botari, T and Galvao, DS},
url = {http://pubs.rsc.org/en/content/articlehtml/2014/cp/c4cp03708a},
year = {2014},
date = {2014-01-01},
journal = {Physical Chemistry Chemical Physics},
volume = {16},
number = {44},
pages = {24570--24574},
publisher = {Royal Society of Chemistry},
abstract = {In this work we report new silicon and germanium tubular nanostructures with no corresponding stable carbon analogues. The electronic and mechanical properties of these new tubes were investigated through ab initio methods. Our results show that these structures have lower energy than their corresponding nanoribbon structures and are stable up to high temperatures (500 and 1000 K, for silicon and germanium tubes, respectively). Both tubes are semiconducting with small indirect band gaps, which can be significantly altered by both compressive and tensile strains. Large bandgap variations of almost 50% were observed for strain rates as small as 3%, suggesting their possible applications in sensor devices. They also present high Young's modulus values (0.25 and 0.15 TPa, respectively). TEM images were simulated to help in the identification of these new structures.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Perim, Eric; Paupitz, Ricardo; Botari, Tiago; Galvao, Douglas S
Novel Semiconducting Silicon and Germanium Nanotubes Journal Article
In: arXiv preprint arXiv:1403.2061, 2014.
@article{perim2014novel,
title = {Novel Semiconducting Silicon and Germanium Nanotubes},
author = {Perim, Eric and Paupitz, Ricardo and Botari, Tiago and Galvao, Douglas S},
url = {http://arxiv.org/abs/1403.2061},
year = {2014},
date = {2014-01-01},
journal = {arXiv preprint arXiv:1403.2061},
abstract = {In this work we report new silicon and germanium nanotube structures, with no corresponding
stable carbon analogues and which cannot be described by integer chiral indices. The electronic
and mechanical properties of these new tubes were investigated through ab initio methods. Our
results show that the structures are stable up to high temperatures (500 and 1000 K, for silicon and
germanium tubes, respectively). Both tubes are semiconducting with small indirect band gaps,
which can be significantly altered by both compressive and tensile strains. They also present high
Young modulus values (0.25 and 0.15 TPa, respectively).},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
stable carbon analogues and which cannot be described by integer chiral indices. The electronic
and mechanical properties of these new tubes were investigated through ab initio methods. Our
results show that the structures are stable up to high temperatures (500 and 1000 K, for silicon and
germanium tubes, respectively). Both tubes are semiconducting with small indirect band gaps,
which can be significantly altered by both compressive and tensile strains. They also present high
Young modulus values (0.25 and 0.15 TPa, respectively).
Perim, Eric; Paupitz, Ricardo; Autreto, PAS; Galvao, DS
The Hydrogenation Dynamics of h-BN Sheets Proceedings
Cambridge University Press, vol. 1549, 2013.
@proceedings{perim2013hydrogenation,
title = {The Hydrogenation Dynamics of h-BN Sheets},
author = {Perim, Eric and Paupitz, Ricardo and Autreto, PAS and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8943477&fileId=S1946427413007938},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1549},
pages = {91--98},
publisher = {Cambridge University Press},
abstract = {Hexagonal boron nitride (h-BN), also known as white graphite, is the inorganic analogue of graphite. Single layers of both structures have been already experimentally realized.
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics of hydrogenation of h-BN single-layers membranes.
Our results show that the rate of hydrogenation atoms bonded to the membrane is highly dependent on the temperature and that only at low temperatures there is a preferential bond to boron atoms. Unlike graphanes (hydrogenated graphene), hydrogenated h-BN membranes do not exhibit the formation of correlated domains. Also, the out-of-plane deformations are more pronounced in comparison with the graphene case. After a critical number of incorporated hydrogen atoms the membrane become increasingly defective, lost its two-dimensional character and collapses. The hydrogen radial pair distribution and second-nearest neighbor correlations were also analyzed.},
keywords = {},
pubstate = {published},
tppubtype = {proceedings}
}
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics of hydrogenation of h-BN single-layers membranes.
Our results show that the rate of hydrogenation atoms bonded to the membrane is highly dependent on the temperature and that only at low temperatures there is a preferential bond to boron atoms. Unlike graphanes (hydrogenated graphene), hydrogenated h-BN membranes do not exhibit the formation of correlated domains. Also, the out-of-plane deformations are more pronounced in comparison with the graphene case. After a critical number of incorporated hydrogen atoms the membrane become increasingly defective, lost its two-dimensional character and collapses. The hydrogen radial pair distribution and second-nearest neighbor correlations were also analyzed.
Garcez, Karl M; Moreira, Edvan; Azevedo, David L; Galvao, Douglas S
Neon atoms oscillating inside carbon and boron nitride nanotubes: a fully atomistic molecular dynamics investigation Journal Article
In: Molecular Simulation, vol. 36, no. 9, pp. 639–643, 2010.
@article{garcez2010neon,
title = {Neon atoms oscillating inside carbon and boron nitride nanotubes: a fully atomistic molecular dynamics investigation},
author = {Garcez, Karl M and Moreira, Edvan and Azevedo, David L and Galvao, Douglas S},
url = {http://www.tandfonline.com/doi/abs/10.1080/08927020903463926#.VLfp54rF-2o},
year = {2010},
date = {2010-01-01},
journal = {Molecular Simulation},
volume = {36},
number = {9},
pages = {639--643},
publisher = {Taylor & Francis Group},
abstract = {In the present work, based on extensive fully atomistic molecular dynamics simulations, we discuss the dynamics of neon atoms oscillating inside (5,5) single-walled carbon nanotubes (CNTs) and boron nitride nanotubes (BNNTs). Our results show that sustained high-frequency oscillatory regimes are possible for a large range of temperatures. Our results also show that the general features of the oscillations are quite similar to those observed in CNT and BNNT, in contrast with some speculations in previous works in the literature about the importance of broken symmetry and chirality exhibited by BNNTs.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Coluci, VR; Braga, SF; Legoas, SB; Galvao, DS; Baughman, RH
New families of carbon nanotubes based on graphyne motifs Journal Article
In: Nanotechnology, vol. 15, no. 4, pp. S142, 2004.
@article{coluci2004new,
title = {New families of carbon nanotubes based on graphyne motifs},
author = {Coluci, VR and Braga, SF and Legoas, SB and Galvao, DS and Baughman, RH},
url = {http://iopscience.iop.org/0957-4484/15/4/006},
year = {2004},
date = {2004-01-01},
journal = {Nanotechnology},
volume = {15},
number = {4},
pages = {S142},
publisher = {IOP Publishing},
abstract = {Electronic properties of proposed new families of carbon single walled nanotubes are investigated. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Analogous to ordinary nanotubes, armchair, zigzag and chiral graphyne nanotubes are possible. Tight-binding and ab initio density functional methods were used to predict the electronic properties of these unusual nanotubes. Of the three graphyne nanotube families analysed here, two provide metallic behaviour for armchair tubes and either metallic or semiconducting behaviour for zigzag nanotubes. For the other graphyne nanotube family investigated a diameter and chirality independent bandgap is predicted and a bandgap modulation study by structural distortions has been carried out for small longitudinal tube deformations. Interestingly, while the bandgap is insensitive to structure, the stress-induced bandgap changes can strongly depend both on the nanotube type and whether the strain is tensile or compressive.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Coluci, VR; Galvao, DS; Baughman, RH
Theoretical investigation of electromechanical effects for graphyne carbon nanotubes Journal Article
In: The Journal of chemical physics, vol. 121, no. 7, pp. 3228–3237, 2004.
@article{coluci2004theoretical,
title = {Theoretical investigation of electromechanical effects for graphyne carbon nanotubes},
author = {Coluci, VR and Galvao, DS and Baughman, RH},
url = {http://scitation.aip.org/content/aip/journal/jcp/121/7/10.1063/1.1772756},
year = {2004},
date = {2004-01-01},
journal = {The Journal of chemical physics},
volume = {121},
number = {7},
pages = {3228--3237},
publisher = {AIP Publishing},
abstract = {We present a theoretical study of the electronic and mechanical properties of graphyne-based nanotubes (GNTs). These semiconducting nanotubes result from the elongation of one-third of the covalent interconnections of graphite-based nanotubes by the introduction of yne groups. The effect of charge injection on the dimensions of GNTs was investigated using tight-binding calculations. Low amounts of electron injection are predicted to cause qualitatively different responses for armchair and zigzag graphyne nanotubes. Although the behavior is qualitatively similar to the usual carbon nanotubes, the charge-induced strains are predicted to be smaller for the GNTs than for ordinary single walled carbon nanotubes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Coluci, VR; Braga, SF; Legoas, SB; Galvao, DS; Baughman, RH
Families of carbon nanotubes: Graphyne-based nanotubes Journal Article
In: Physical Review B, vol. 68, no. 3, pp. 035430, 2003.
@article{coluci2003families,
title = {Families of carbon nanotubes: Graphyne-based nanotubes},
author = {Coluci, VR and Braga, SF and Legoas, SB and Galvao, DS and Baughman, RH},
url = {http://journals.aps.org/prb/abstract/10.1103/PhysRevB.68.035430},
year = {2003},
date = {2003-01-01},
journal = {Physical Review B},
volume = {68},
number = {3},
pages = {035430},
publisher = {APS},
abstract = {New families of carbon single-walled nanotubes are proposed and their electronic structures are investigated. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Analogously to ordinary nanotubes, armchair, zigzag, and chiral graphyne nanotubes are possible. We here predict the electronic properties of these unusual nanotubes using tight-binding and ab initio density functional methods. Of the three graphyne nanotube families analyzed here, two provide metallic behavior for armchair tubes and either metallic or semiconducting behavior for zigzag nanotubes. A diameter- and chirality-independent band gap is predicted for the other investigated graphyne family, as well as an oscillatory dependence of the effective mass on nanotube diameter.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Coluci, VR; Braga, SF; Legoas, SB; Galvao, DS; Baughman, RH
Graphyne Nanotubes: New Families of Carbon Nanotubes Proceedings
Warrendale, Pa.; Materials Research Society; 1999, vol. 739, 2003.
@proceedings{coluci2003graphyne,
title = {Graphyne Nanotubes: New Families of Carbon Nanotubes},
author = {Coluci, VR and Braga, SF and Legoas, SB and Galvao, DS and Baughman, RH},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8031794},
year = {2003},
date = {2003-01-01},
journal = {MATERIALS RESEARCH SOCIETY SYMPOSIUM PROCEEDINGS},
volume = {739},
pages = {175--180},
publisher = {Warrendale, Pa.; Materials Research Society; 1999},
abstract = {Fundamentally new families of carbon single walled nanotubes are proposed. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Similarly to ordinary nanotubes, armchair, zig-zag, and chiral graphyne nanotubes are possible. We present here results for the electronic properties of graphyne based tubes obtained from tight-binding and ab initio density functional methods.},
keywords = {},
pubstate = {published},
tppubtype = {proceedings}
}
Coluci, V R; Braga, SF; Legoas, Sergio B; Galvao, Douglas S; Baughman, RH
New families of carbon nanotubes Journal Article
In: arXiv preprint cond-mat/0207085, 2002.
@article{coluci2002new,
title = {New families of carbon nanotubes},
author = {Coluci, V R and Braga, SF and Legoas, Sergio B and Galvao, Douglas S and Baughman, RH},
url = {http://arxiv.org/abs/cond-mat/0207085},
year = {2002},
date = {2002-01-01},
journal = {arXiv preprint cond-mat/0207085},
abstract = {Fundamentally new families of carbon single walled nanotubes are proposed. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Similarly to ordinary nanotubes, arm-chair, zig-zag, and chiral graphyne nanotubes are possible. Electronic properties, predicted using tight-binding and ab initio density functional methods, show a rich variety of metallic and semiconducting behaviors.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2019
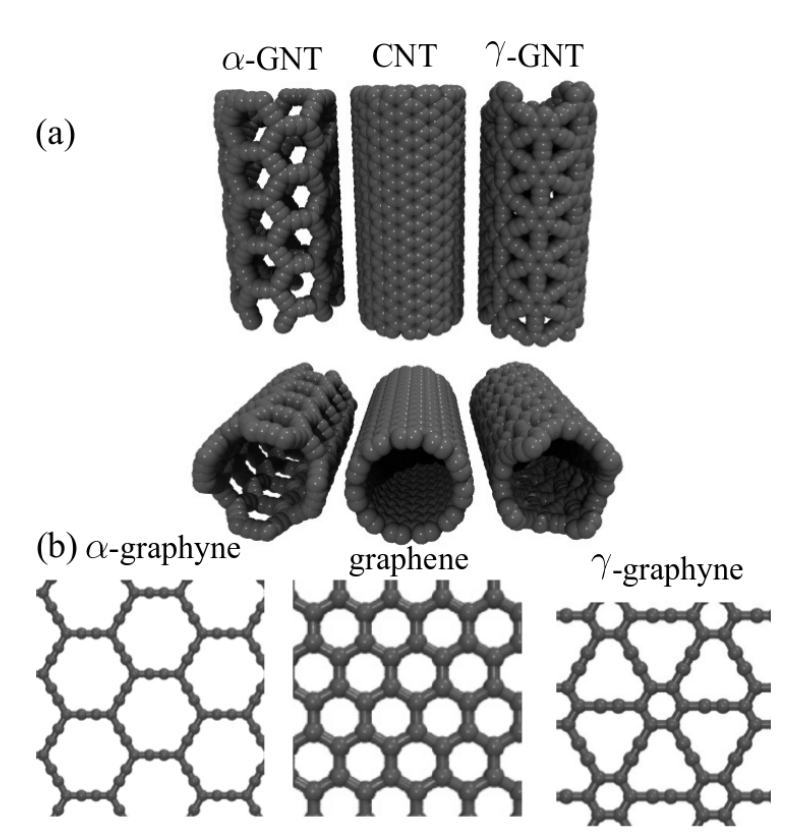
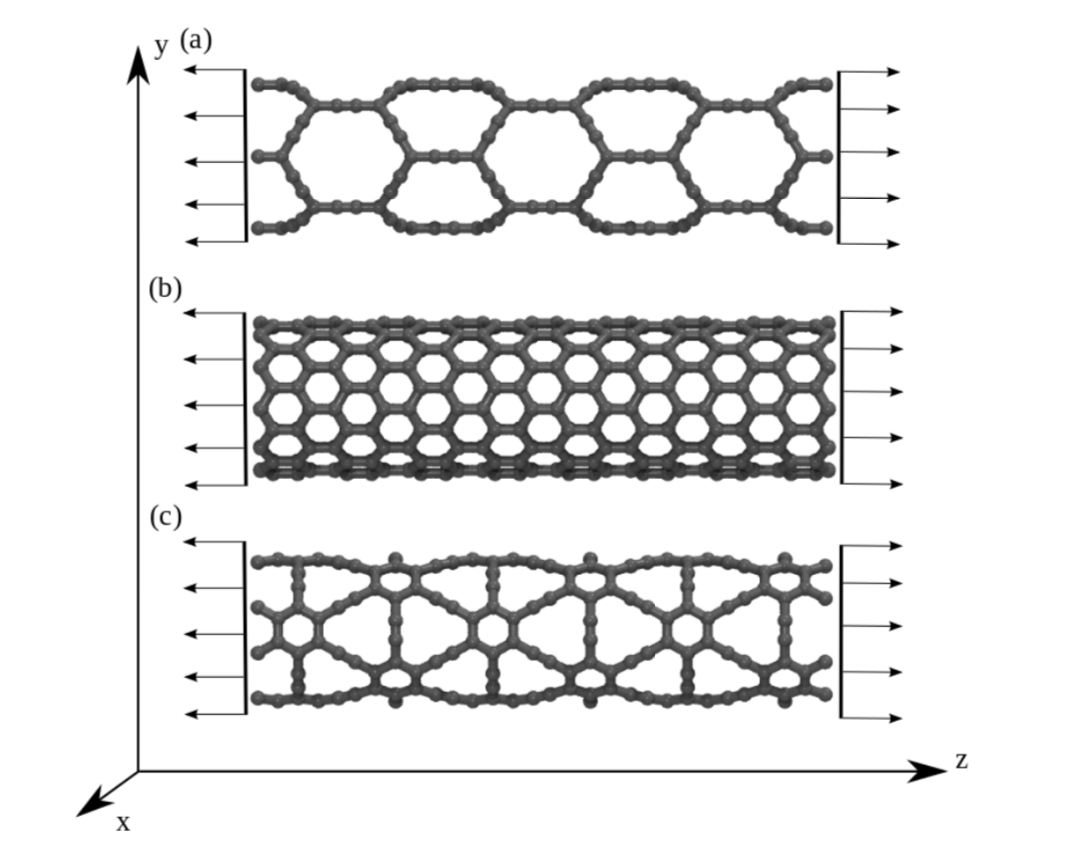
JM; Sousa, Bizao
Elastic Properties of Graphyne-based Nanotubes Online
2019, (ArXiv preprint.).
Abstract | Links | BibTeX | Tags: DFT, Graphynes, Molecular Dynamics, Nanotubes
@online{deSousa2019b,
title = {Elastic Properties of Graphyne-based Nanotubes},
author = {de Sousa, JM; , Bizao, RA; Sousa Filho, VP; Aguiar, AL; Coluci, VR; Pugno, NM; Girao, EC; Souza Filho, AG; Galvao, DS},
url = {https://arxiv.org/pdf/1905.02104.pdf},
year = {2019},
date = {2019-04-07},
abstract = {Graphyne nanotubes (GNTs) are nanostructures obtained from rolled up graphyne sheets,
in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes
are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to
conventional CNTs, GNTs can present different chiralities and electronic properties. Because
of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their
mechanical properties. In this work, we studied the mechanical response of GNTs under
tensile stress using fully atomistic molecular dynamics simulations and density functional
theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs
at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller
ultimate strength and Young’s modulus values. This is a consequence of the combined
effects of the existence of triple bonds and increased porosity/flexibility due to the presence
of acetylenic groups.},
note = {ArXiv preprint.},
keywords = {DFT, Graphynes, Molecular Dynamics, Nanotubes},
pubstate = {published},
tppubtype = {online}
}
in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes
are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to
conventional CNTs, GNTs can present different chiralities and electronic properties. Because
of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their
mechanical properties. In this work, we studied the mechanical response of GNTs under
tensile stress using fully atomistic molecular dynamics simulations and density functional
theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs
at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller
ultimate strength and Young’s modulus values. This is a consequence of the combined
effects of the existence of triple bonds and increased porosity/flexibility due to the presence
of acetylenic groups.
JM; Sousa, Bizao
Elastic Properties of Graphyne-Based Nanotubes Journal Article
In: Computational Materials Science, vol. 170, pp. 109153, 2019.
Abstract | Links | BibTeX | Tags: DFT, Graphynes, Molecular Dynamics, Nanotubes
@article{deSousa2019c,
title = {Elastic Properties of Graphyne-Based Nanotubes},
author = {de Sousa, JM; , Bizao, RA; Sousa Filho, VP; Aguiar, AL; Coluci, VR; Pugno, NM; Girao, EC; Souza Filho, AG; Galvao, DS},
url = {https://www.sciencedirect.com/science/article/pii/S0927025619304525?dgcid=coauthor#s0040},
doi = {10.1016/j.commatsci.2019.109153},
year = {2019},
date = {2019-04-03},
journal = {Computational Materials Science},
volume = {170},
pages = {109153},
abstract = {Graphyne nanotubes (GNTs) are nanostructures obtained from rolled up graphyne sheets, in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to conventional CNTs, GNTs can present different chiralities and electronic properties. Because of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their mechanical properties. In this work, we studied the mechanical response of GNTs under tensile stress using fully atomistic molecular dynamics simulations and density functional theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller ultimate strength and Young’s modulus values. This is a consequence of the combined effects of the existence of triple bonds and increased porosity/flexibility due to the presence of acetylenic groups.},
keywords = {DFT, Graphynes, Molecular Dynamics, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
2018
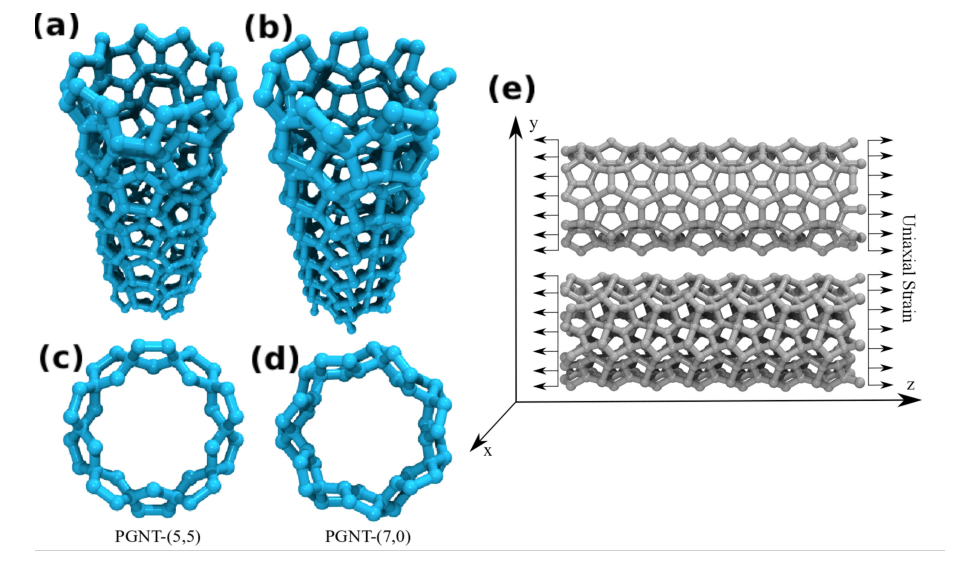
de Sousa, J. M.; Aguiar, A. L.; Girao, E. C.; Fonseca, Alexandre F.; Filho, A. G. Sousa; Galvao, Douglas S.
Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study Online
2018, (preprint arXiv:1801.04269).
Abstract | Links | BibTeX | Tags: Fracture, Molecular Dynamics, Nanotubes, pentagraphene
@online{deSousa2018d,
title = {Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study},
author = {J. M. de Sousa and A. L. Aguiar and E. C. Girao and Alexandre F. Fonseca and A. G. Sousa Filho and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.04269},
year = {2018},
date = {2018-01-15},
abstract = {The study of the mechanical properties of nanostructured systems has gained importance in
theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of
the strongest nanomaterials found in nature, with Young's Modulus (YM) in the order 1.25
TPa. One interesting question is about the possibility of generating new nanostructures with
1D symmetry and with similar and/or superior CNT properties. In this work, we present a
study on the dynamical, structural, mechanical properties, fracture patterns and YM values
for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These
tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional
structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized
states) in the same form that CNTs are formed from rolling up graphene membranes. We
carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We
have considered zigzag-like and armchair-like PGNTs of different diameters. Our results
show that PGNTs present YM ~ 800 GPa with distinct elastic behavior in relation to CNTs,
mainly associated with mechanical failure, chirality dependent fracture patterns and extensive
structural reconstructions},
note = {preprint arXiv:1801.04269},
keywords = {Fracture, Molecular Dynamics, Nanotubes, pentagraphene},
pubstate = {published},
tppubtype = {online}
}
theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of
the strongest nanomaterials found in nature, with Young's Modulus (YM) in the order 1.25
TPa. One interesting question is about the possibility of generating new nanostructures with
1D symmetry and with similar and/or superior CNT properties. In this work, we present a
study on the dynamical, structural, mechanical properties, fracture patterns and YM values
for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These
tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional
structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized
states) in the same form that CNTs are formed from rolling up graphene membranes. We
carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We
have considered zigzag-like and armchair-like PGNTs of different diameters. Our results
show that PGNTs present YM ~ 800 GPa with distinct elastic behavior in relation to CNTs,
mainly associated with mechanical failure, chirality dependent fracture patterns and extensive
structural reconstructions
2016
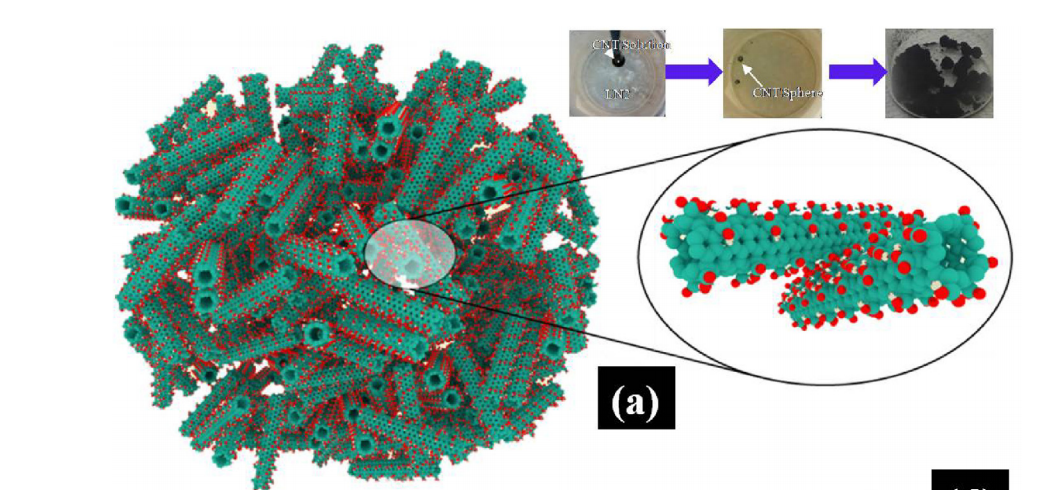
Amelia HC Hart Ryota Koizumi, Gustavo Brunetto
Mechano-chemical stabilization of three-dimensional carbon nanotube aggregates Journal Article
In: Carbon, vol. 110, pp. 27-33, 2016.
Abstract | Links | BibTeX | Tags: Mechano-chemistry, Molecular Dynamics, Nanotubes
@article{koizumi2016mechano,
title = {Mechano-chemical stabilization of three-dimensional carbon nanotube aggregates},
author = {Ryota Koizumi, Amelia HC Hart, Gustavo Brunetto, Sanjit Bhowmick, Peter S Owuor, John T Hamel, Anieph X Gentles, Sehmus Ozden, Jun Lou, Robert Vajtai, SA Syed Asif, Douglas S Galvão, CS Tiwary, PM Ajayan},
url = {www.sciencedirect.com/science/article/pii/S0008622316307400},
doi = {10.1016/j.carbon.2016.08.085},
year = {2016},
date = {2016-08-21},
journal = {Carbon},
volume = {110},
pages = {27-33},
abstract = {Here we report a combined study of experiments and simulations to understand how chemical functional groups can mechanically stabilize aggregates of carbon nanotubes (CNTs). Ultralow density aggregates of chemically functionalized CNTs, in the form of macro-scale spheres made by freeze-drying method, show mechanical stabilization and near complete elastic recovery during deformation. Simulations of interacting functionalized carbon nanotube aggregates show better structural retention compared to non-functionalized CNTs under compression, suggesting that the atomic-level interactions between functional groups on adjoining CNTs help maintain structural rigidity and elastic response during loading. Aggregates of non-functionalized CNTs collapses under similar loading conditions. The dynamic mechanical responses of CNT macrostructures and mechano-chemical stabilization are directly observed using in-situ deformation inside a scanning electron microscope.},
keywords = {Mechano-chemistry, Molecular Dynamics, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
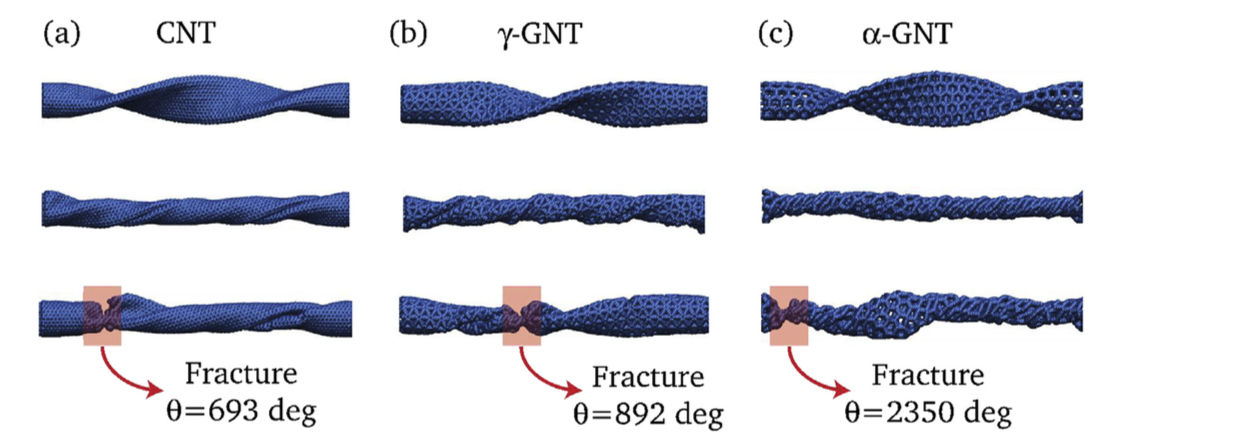
G. Brunetto J.M. de Sousa, V. R. Coluci
Torsional “superplasticity” of graphyne nanotubes Journal Article
In: Carbon, vol. 96, pp. 14-19, 2016.
Abstract | Links | BibTeX | Tags: Fracture, Graphynes, Mechanical Properties, Nanotubes
@article{deSousa2016,
title = {Torsional “superplasticity” of graphyne nanotubes},
author = {J.M. de Sousa, G. Brunetto, V.R. Coluci, D.S. Galvao },
url = {http://www.sciencedirect.com/science/article/pii/S000862231530258X},
doi = { http://dx.doi.org/10.1016/j.carbon.2015.09.039},
year = {2016},
date = {2016-01-01},
journal = {Carbon},
volume = {96},
pages = {14-19},
abstract = {Graphyne is a planar two-dimensional carbon allotrope formed by atoms in sp, sp2, and sp3 hybridized states. Topologically graphyne nanotubes (GNTs) can be considered as cylindrically rolled up graphyne sheets, similarly as carbon nanotubes (CNTs) can be considered rolled up graphene sheets. Due to the presence of single, double, and triple bonds, GNTs exhibit porous sidewalls that can be exploited in many diverse applications. In this work, we investigated the mechanical behavior of GNTs under torsional strains through reactive molecular dynamics simulations. Our results show that GNTs are more flexible than CNTs and exhibit “superplasticit”, with fracture angles that are up to 35 times higher than the ones reported to CNTs. This GNT “superplastic” behavior can be explained in terms of irreversible recon- struction processes (mainly associated with the triple bonds) that occur during torsional strains.},
keywords = {Fracture, Graphynes, Mechanical Properties, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
2015
Gustavo Brunetto Jose M. de Sousa, Vitor R. Coluci
Torsional "Superplasticity" of Graphyne Nanotubes Online
2015, (ArXiv reprint of Torsional "Superplasticity" of Graphyne Nanotubes, published in Carbon 96, 14 (2016).).
Abstract | Links | BibTeX | Tags: Allotropes, Graphynes, Mechanical Properties, Nanotubes
@online{deSousa2015b,
title = {Torsional "Superplasticity" of Graphyne Nanotubes},
author = {Jose M. de Sousa, Gustavo Brunetto, Vitor R. Coluci, Douglas S. Galvao},
url = {http://arxiv.org/abs/1509.08746},
year = {2015},
date = {2015-09-29},
abstract = {Graphyne is a planar two-dimensional carbon allotrope formed by atoms in sp, sp2, and sp3 hybridized states. Topologically graphyne nanotubes (GNTs) can be considered as cylindrically rolled up graphyne sheets, similarly as carbon nanotubes (CNTs) can be considered rolled up graphene sheets. Due to the presence of single, double, and triple bonds, GNTs exhibit porous sidewalls that can be exploited in many diverse applications. In this work, we investigated the mechanical behavior of GNTs under torsional strains through reactive molecular dynamics simulations. Our results show that GNTs are more flexible than CNTs and exhibit 'superplasticity', with fracture angles that are up to 35 times higher than the ones reported to CNTs. This GNT 'superplastic' behavior can be explained in terms of irreversible reconstruction processes (mainly associated with the triple bonds) that occur during torsional strains.},
note = {ArXiv reprint of Torsional "Superplasticity" of Graphyne Nanotubes, published in Carbon 96, 14 (2016).},
keywords = {Allotropes, Graphynes, Mechanical Properties, Nanotubes},
pubstate = {published},
tppubtype = {online}
}
2014
Perim, E; Paupitz, R; Botari, T; Galvao, DS
One-dimensional silicon and germanium nanostructures with no carbon analogues Journal Article
In: Physical Chemistry Chemical Physics, vol. 16, no. 44, pp. 24570–24574, 2014.
Abstract | Links | BibTeX | Tags: DFT, Germanium, Nanotubes, Silicon
@article{perim2014one,
title = {One-dimensional silicon and germanium nanostructures with no carbon analogues},
author = {Perim, E and Paupitz, R and Botari, T and Galvao, DS},
url = {http://pubs.rsc.org/en/content/articlehtml/2014/cp/c4cp03708a},
year = {2014},
date = {2014-01-01},
journal = {Physical Chemistry Chemical Physics},
volume = {16},
number = {44},
pages = {24570--24574},
publisher = {Royal Society of Chemistry},
abstract = {In this work we report new silicon and germanium tubular nanostructures with no corresponding stable carbon analogues. The electronic and mechanical properties of these new tubes were investigated through ab initio methods. Our results show that these structures have lower energy than their corresponding nanoribbon structures and are stable up to high temperatures (500 and 1000 K, for silicon and germanium tubes, respectively). Both tubes are semiconducting with small indirect band gaps, which can be significantly altered by both compressive and tensile strains. Large bandgap variations of almost 50% were observed for strain rates as small as 3%, suggesting their possible applications in sensor devices. They also present high Young's modulus values (0.25 and 0.15 TPa, respectively). TEM images were simulated to help in the identification of these new structures.},
keywords = {DFT, Germanium, Nanotubes, Silicon},
pubstate = {published},
tppubtype = {article}
}
Perim, Eric; Paupitz, Ricardo; Botari, Tiago; Galvao, Douglas S
Novel Semiconducting Silicon and Germanium Nanotubes Journal Article
In: arXiv preprint arXiv:1403.2061, 2014.
Abstract | Links | BibTeX | Tags: DFT, Germanium, Nanotubes, Silicon
@article{perim2014novel,
title = {Novel Semiconducting Silicon and Germanium Nanotubes},
author = {Perim, Eric and Paupitz, Ricardo and Botari, Tiago and Galvao, Douglas S},
url = {http://arxiv.org/abs/1403.2061},
year = {2014},
date = {2014-01-01},
journal = {arXiv preprint arXiv:1403.2061},
abstract = {In this work we report new silicon and germanium nanotube structures, with no corresponding
stable carbon analogues and which cannot be described by integer chiral indices. The electronic
and mechanical properties of these new tubes were investigated through ab initio methods. Our
results show that the structures are stable up to high temperatures (500 and 1000 K, for silicon and
germanium tubes, respectively). Both tubes are semiconducting with small indirect band gaps,
which can be significantly altered by both compressive and tensile strains. They also present high
Young modulus values (0.25 and 0.15 TPa, respectively).},
keywords = {DFT, Germanium, Nanotubes, Silicon},
pubstate = {published},
tppubtype = {article}
}
stable carbon analogues and which cannot be described by integer chiral indices. The electronic
and mechanical properties of these new tubes were investigated through ab initio methods. Our
results show that the structures are stable up to high temperatures (500 and 1000 K, for silicon and
germanium tubes, respectively). Both tubes are semiconducting with small indirect band gaps,
which can be significantly altered by both compressive and tensile strains. They also present high
Young modulus values (0.25 and 0.15 TPa, respectively).
2013
Perim, Eric; Paupitz, Ricardo; Autreto, PAS; Galvao, DS
The Hydrogenation Dynamics of h-BN Sheets Proceedings
Cambridge University Press, vol. 1549, 2013.
Abstract | Links | BibTeX | Tags: Boron Nitride, Hydrogenation, Molecular Dynamics, Nanotubes
@proceedings{perim2013hydrogenation,
title = {The Hydrogenation Dynamics of h-BN Sheets},
author = {Perim, Eric and Paupitz, Ricardo and Autreto, PAS and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8943477&fileId=S1946427413007938},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1549},
pages = {91--98},
publisher = {Cambridge University Press},
abstract = {Hexagonal boron nitride (h-BN), also known as white graphite, is the inorganic analogue of graphite. Single layers of both structures have been already experimentally realized.
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics of hydrogenation of h-BN single-layers membranes.
Our results show that the rate of hydrogenation atoms bonded to the membrane is highly dependent on the temperature and that only at low temperatures there is a preferential bond to boron atoms. Unlike graphanes (hydrogenated graphene), hydrogenated h-BN membranes do not exhibit the formation of correlated domains. Also, the out-of-plane deformations are more pronounced in comparison with the graphene case. After a critical number of incorporated hydrogen atoms the membrane become increasingly defective, lost its two-dimensional character and collapses. The hydrogen radial pair distribution and second-nearest neighbor correlations were also analyzed.},
keywords = {Boron Nitride, Hydrogenation, Molecular Dynamics, Nanotubes},
pubstate = {published},
tppubtype = {proceedings}
}
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics of hydrogenation of h-BN single-layers membranes.
Our results show that the rate of hydrogenation atoms bonded to the membrane is highly dependent on the temperature and that only at low temperatures there is a preferential bond to boron atoms. Unlike graphanes (hydrogenated graphene), hydrogenated h-BN membranes do not exhibit the formation of correlated domains. Also, the out-of-plane deformations are more pronounced in comparison with the graphene case. After a critical number of incorporated hydrogen atoms the membrane become increasingly defective, lost its two-dimensional character and collapses. The hydrogen radial pair distribution and second-nearest neighbor correlations were also analyzed.
2010
Garcez, Karl M; Moreira, Edvan; Azevedo, David L; Galvao, Douglas S
Neon atoms oscillating inside carbon and boron nitride nanotubes: a fully atomistic molecular dynamics investigation Journal Article
In: Molecular Simulation, vol. 36, no. 9, pp. 639–643, 2010.
Abstract | Links | BibTeX | Tags: Boron Nitride, Encapsulation, Molecular Dynamics, Nanotubes
@article{garcez2010neon,
title = {Neon atoms oscillating inside carbon and boron nitride nanotubes: a fully atomistic molecular dynamics investigation},
author = {Garcez, Karl M and Moreira, Edvan and Azevedo, David L and Galvao, Douglas S},
url = {http://www.tandfonline.com/doi/abs/10.1080/08927020903463926#.VLfp54rF-2o},
year = {2010},
date = {2010-01-01},
journal = {Molecular Simulation},
volume = {36},
number = {9},
pages = {639--643},
publisher = {Taylor & Francis Group},
abstract = {In the present work, based on extensive fully atomistic molecular dynamics simulations, we discuss the dynamics of neon atoms oscillating inside (5,5) single-walled carbon nanotubes (CNTs) and boron nitride nanotubes (BNNTs). Our results show that sustained high-frequency oscillatory regimes are possible for a large range of temperatures. Our results also show that the general features of the oscillations are quite similar to those observed in CNT and BNNT, in contrast with some speculations in previous works in the literature about the importance of broken symmetry and chirality exhibited by BNNTs.},
keywords = {Boron Nitride, Encapsulation, Molecular Dynamics, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
2004
Coluci, VR; Braga, SF; Legoas, SB; Galvao, DS; Baughman, RH
New families of carbon nanotubes based on graphyne motifs Journal Article
In: Nanotechnology, vol. 15, no. 4, pp. S142, 2004.
Abstract | Links | BibTeX | Tags: Allotropes, Electronic Structure, Graphynes, Nanotubes
@article{coluci2004new,
title = {New families of carbon nanotubes based on graphyne motifs},
author = {Coluci, VR and Braga, SF and Legoas, SB and Galvao, DS and Baughman, RH},
url = {http://iopscience.iop.org/0957-4484/15/4/006},
year = {2004},
date = {2004-01-01},
journal = {Nanotechnology},
volume = {15},
number = {4},
pages = {S142},
publisher = {IOP Publishing},
abstract = {Electronic properties of proposed new families of carbon single walled nanotubes are investigated. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Analogous to ordinary nanotubes, armchair, zigzag and chiral graphyne nanotubes are possible. Tight-binding and ab initio density functional methods were used to predict the electronic properties of these unusual nanotubes. Of the three graphyne nanotube families analysed here, two provide metallic behaviour for armchair tubes and either metallic or semiconducting behaviour for zigzag nanotubes. For the other graphyne nanotube family investigated a diameter and chirality independent bandgap is predicted and a bandgap modulation study by structural distortions has been carried out for small longitudinal tube deformations. Interestingly, while the bandgap is insensitive to structure, the stress-induced bandgap changes can strongly depend both on the nanotube type and whether the strain is tensile or compressive.
},
keywords = {Allotropes, Electronic Structure, Graphynes, Nanotubes},
pubstate = {published},
tppubtype = {article}
}

Coluci, VR; Galvao, DS; Baughman, RH
Theoretical investigation of electromechanical effects for graphyne carbon nanotubes Journal Article
In: The Journal of chemical physics, vol. 121, no. 7, pp. 3228–3237, 2004.
Abstract | Links | BibTeX | Tags: Allotropes, Electroactuation, Electronic Structure, Graphynes, Nanotubes
@article{coluci2004theoretical,
title = {Theoretical investigation of electromechanical effects for graphyne carbon nanotubes},
author = {Coluci, VR and Galvao, DS and Baughman, RH},
url = {http://scitation.aip.org/content/aip/journal/jcp/121/7/10.1063/1.1772756},
year = {2004},
date = {2004-01-01},
journal = {The Journal of chemical physics},
volume = {121},
number = {7},
pages = {3228--3237},
publisher = {AIP Publishing},
abstract = {We present a theoretical study of the electronic and mechanical properties of graphyne-based nanotubes (GNTs). These semiconducting nanotubes result from the elongation of one-third of the covalent interconnections of graphite-based nanotubes by the introduction of yne groups. The effect of charge injection on the dimensions of GNTs was investigated using tight-binding calculations. Low amounts of electron injection are predicted to cause qualitatively different responses for armchair and zigzag graphyne nanotubes. Although the behavior is qualitatively similar to the usual carbon nanotubes, the charge-induced strains are predicted to be smaller for the GNTs than for ordinary single walled carbon nanotubes.},
keywords = {Allotropes, Electroactuation, Electronic Structure, Graphynes, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
2003
Coluci, VR; Braga, SF; Legoas, SB; Galvao, DS; Baughman, RH
Families of carbon nanotubes: Graphyne-based nanotubes Journal Article
In: Physical Review B, vol. 68, no. 3, pp. 035430, 2003.
Abstract | Links | BibTeX | Tags: Allotropes, Electronic Structure, Graphynes, Nanotubes
@article{coluci2003families,
title = {Families of carbon nanotubes: Graphyne-based nanotubes},
author = {Coluci, VR and Braga, SF and Legoas, SB and Galvao, DS and Baughman, RH},
url = {http://journals.aps.org/prb/abstract/10.1103/PhysRevB.68.035430},
year = {2003},
date = {2003-01-01},
journal = {Physical Review B},
volume = {68},
number = {3},
pages = {035430},
publisher = {APS},
abstract = {New families of carbon single-walled nanotubes are proposed and their electronic structures are investigated. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Analogously to ordinary nanotubes, armchair, zigzag, and chiral graphyne nanotubes are possible. We here predict the electronic properties of these unusual nanotubes using tight-binding and ab initio density functional methods. Of the three graphyne nanotube families analyzed here, two provide metallic behavior for armchair tubes and either metallic or semiconducting behavior for zigzag nanotubes. A diameter- and chirality-independent band gap is predicted for the other investigated graphyne family, as well as an oscillatory dependence of the effective mass on nanotube diameter.},
keywords = {Allotropes, Electronic Structure, Graphynes, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
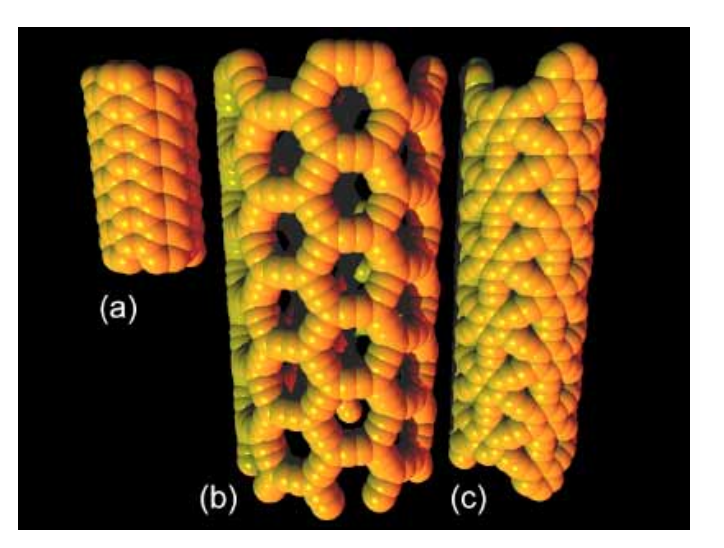
Coluci, VR; Braga, SF; Legoas, SB; Galvao, DS; Baughman, RH
Graphyne Nanotubes: New Families of Carbon Nanotubes Proceedings
Warrendale, Pa.; Materials Research Society; 1999, vol. 739, 2003.
Abstract | Links | BibTeX | Tags: Allotropes, Electronic Structure, Graphynes, Nanotubes
@proceedings{coluci2003graphyne,
title = {Graphyne Nanotubes: New Families of Carbon Nanotubes},
author = {Coluci, VR and Braga, SF and Legoas, SB and Galvao, DS and Baughman, RH},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8031794},
year = {2003},
date = {2003-01-01},
journal = {MATERIALS RESEARCH SOCIETY SYMPOSIUM PROCEEDINGS},
volume = {739},
pages = {175--180},
publisher = {Warrendale, Pa.; Materials Research Society; 1999},
abstract = {Fundamentally new families of carbon single walled nanotubes are proposed. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Similarly to ordinary nanotubes, armchair, zig-zag, and chiral graphyne nanotubes are possible. We present here results for the electronic properties of graphyne based tubes obtained from tight-binding and ab initio density functional methods.},
keywords = {Allotropes, Electronic Structure, Graphynes, Nanotubes},
pubstate = {published},
tppubtype = {proceedings}
}
2002
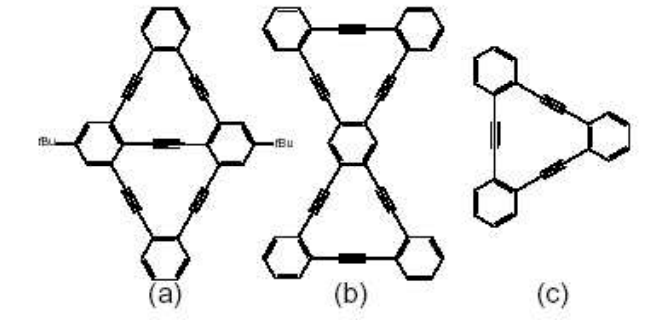
Coluci, V R; Braga, SF; Legoas, Sergio B; Galvao, Douglas S; Baughman, RH
New families of carbon nanotubes Journal Article
In: arXiv preprint cond-mat/0207085, 2002.
Abstract | Links | BibTeX | Tags: Allotropes, Electronic Structure, Graphynes, Nanotubes
@article{coluci2002new,
title = {New families of carbon nanotubes},
author = {Coluci, V R and Braga, SF and Legoas, Sergio B and Galvao, Douglas S and Baughman, RH},
url = {http://arxiv.org/abs/cond-mat/0207085},
year = {2002},
date = {2002-01-01},
journal = {arXiv preprint cond-mat/0207085},
abstract = {Fundamentally new families of carbon single walled nanotubes are proposed. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Similarly to ordinary nanotubes, arm-chair, zig-zag, and chiral graphyne nanotubes are possible. Electronic properties, predicted using tight-binding and ab initio density functional methods, show a rich variety of metallic and semiconducting behaviors.},
keywords = {Allotropes, Electronic Structure, Graphynes, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ

