
Chandra Sekhar Tiwary Dibyendu Chakravarty, Leonardo Dantas Machado
Zirconia-Nanoparticle-Reinforced Morphology-Engineered Graphene-Based Foams Journal Article
In: Advanced Materials, vol. 27, no. 31, pp. 4534–4543, 2015.
@article{Chakravarty2015,
title = {Zirconia-Nanoparticle-Reinforced Morphology-Engineered Graphene-Based Foams},
author = { Dibyendu Chakravarty , Chandra Sekhar Tiwary , Leonardo Dantas Machado ,
Gustavo Brunetto , Soumya Vinod , Ram Manohar Yadav , Douglas S. Galvao ,
Shrikant V. Joshi , Govindan Sundararajan, Pulickel M. Ajayan },
url = {http://onlinelibrary.wiley.com/doi/10.1002/adma.201502409/full},
doi = {10.1002/adma.201502409},
year = {2015},
date = {2015-07-15},
journal = {Advanced Materials},
volume = {27},
number = {31},
pages = {4534–4543},
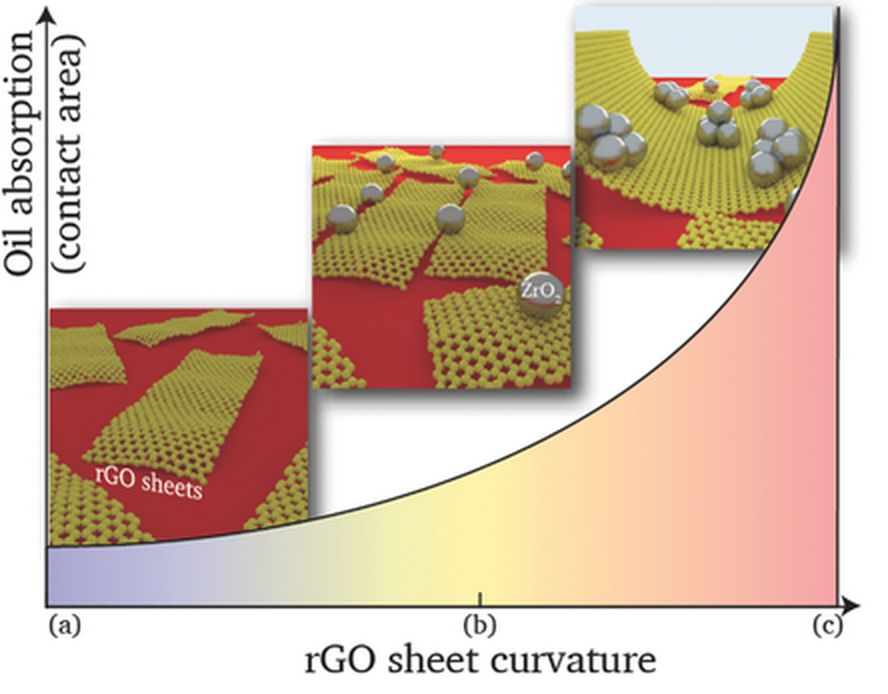
abstract = {The morphology of graphene-based foams can be engineered by reinforcing them with nanocrystalline zirconia, thus improving their oil-adsorption capacity; This can be observed experimentally and explained theoretically. Low zirconia fractions yield flaky microstructures where zirconia nanoparticles arrest propagating cracks. Higher zirconia concentrations possess a mesh-like interconnected structure where the degree of coiling is dependant on the local zirconia content.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Yongji Gong Kunttal Keyshar, Gonglan Ye
Chemical Vapor Deposition of Monolayer Rhenium Disulfide (ReS2) Journal Article
In: Advanced Materials, vol. 27, no. 31, pp. 4640–4648, 2015.
@article{Keyshar2015,
title = {Chemical Vapor Deposition of Monolayer Rhenium Disulfide (ReS2)},
author = {Kunttal Keyshar , Yongji Gong , Gonglan Ye , Gustavo Brunetto , Wu Zhou ,
Daniel P. Cole , Ken Hackenberg , Yongmin He , Leonardo Machado , Mohamad Kabbani ,
Amelia H. C. Hart , Bo Li , Douglas S. Galvao , Antony George , Robert Vajtai ,
Chandra Sekhar Tiwary , Pulickel M. Ajayan},
url = {http://onlinelibrary.wiley.com/doi/10.1002/adma.201501795/full},
doi = {10.1002/adma.201501795},
year = {2015},
date = {2015-07-03},
journal = {Advanced Materials},
volume = {27},
number = {31},
pages = {4640–4648},
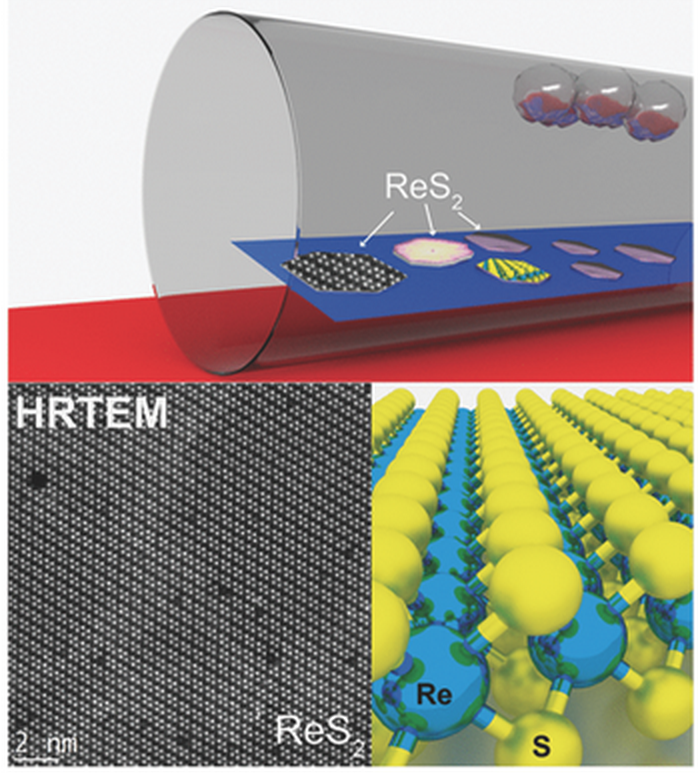
abstract = {The direct synthesis of monolayer and multilayer ReS2 by chemical vapor deposition at a low temperature of 450 °C is reported. Detailed characterization of this material is performed using various spectroscopy and microscopy methods. Furthermore initial field-effect transistor characteristics are evaluated, which highlight the potential in being used as an n-type semiconductor.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Chandra Sekhar Tiwary Mohamad A Kabbani, Pedro AS Autreto
Ambient solid-state mechano-chemical reactions between functionalized carbon nanotubes Journal Article
In: Nature Communications, vol. 6, pp. 7291, 2015.
@article{Kabbani2015,
title = {Ambient solid-state mechano-chemical reactions between functionalized carbon nanotubes},
author = {Mohamad A Kabbani, Chandra Sekhar Tiwary, Pedro AS Autreto, Gustavo Brunetto, Anirban Som, KR Krishnadas, Sehmus Ozden, Ken P Hackenberg, Yongi Gong, Douglas S Galvao, Robert Vajtai, Ahmad T Kabbani, Thalappil Pradeep, Pulickel M Ajayan},
url = {http://www.nature.com/ncomms/2015/150615/ncomms8291/full/ncomms8291.html},
doi = {10.1038/ncomms8291},
year = {2015},
date = {2015-06-15},
journal = {Nature Communications},
volume = {6},
pages = {7291},
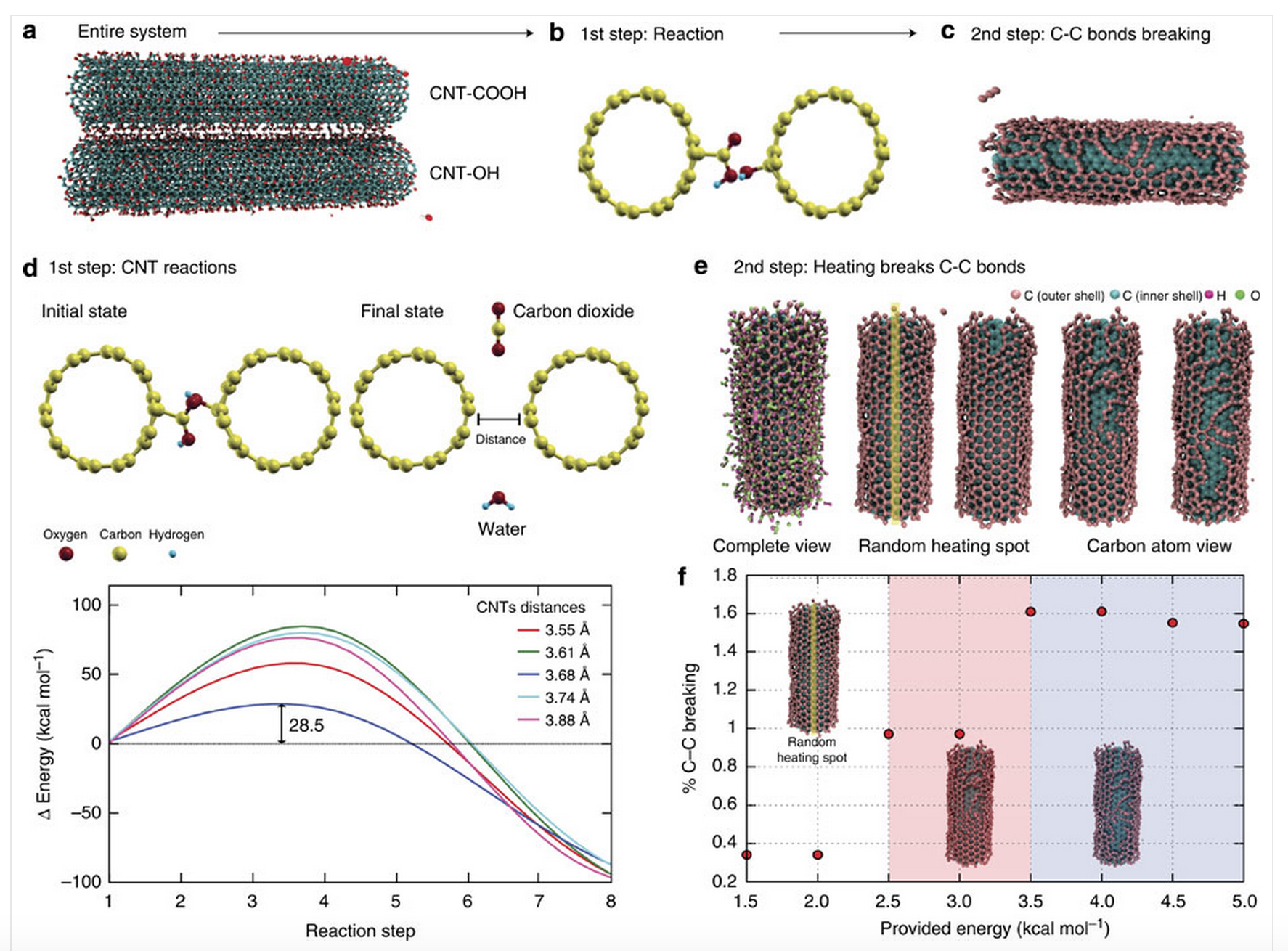
abstract = {Carbon nanotubes can be chemically modified by attaching various functionalities to their surfaces, although harsh chemical treatments can lead to their break-up into graphene nanostructures. On the other hand, direct coupling between functionalities bound on individual nanotubes could lead to, as yet unexplored, spontaneous chemical reactions. Here we report an ambient mechano-chemical reaction between two varieties of nanotubes, carrying predominantly carboxyl and hydroxyl functionalities, respectively, facilitated by simple mechanical grinding of the reactants. The purely solid-state reaction between the chemically differentiated nanotube species produces condensation products and unzipping of nanotubes due to local energy release, as confirmed by spectroscopic measurements, thermal analysis and molecular dynamic simulations.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Acrísio L Aguiar Nadia Ferreira Andrade, Yoong Ahm Kim
Linear Carbon Chains Under High Pressure Conditions Journal Article
In: The Journal of Physical Chemistry C, vol. 119, no. 19, pp. 10669–10676, 2015.
@article{Andrade2015,
title = {Linear Carbon Chains Under High Pressure Conditions},
author = {Nadia Ferreira Andrade, Acrísio L Aguiar, Yoong Ahm Kim, Morinobu Endo, Paulo TC Freire, Gustavo Bruneto, Douglas Soares Galvao, Mildred S Dresselhaus, Antonio Gomes Souza Filho},
url = {http://pubs.acs.org/doi/abs/10.1021/acs.jpcc.5b00902},
doi = {10.1021/acs.jpcc.5b00902},
year = {2015},
date = {2015-04-23},
journal = {The Journal of Physical Chemistry C},
volume = {119},
number = {19},
pages = {10669–10676},
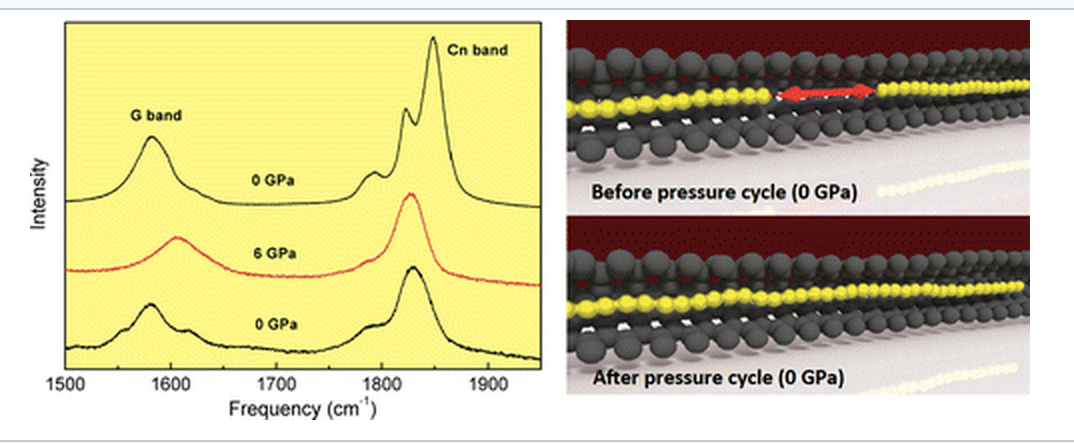
abstract = {A high-pressure resonance Raman spectroscopy study of linear carbon chains encapsulated inside multiwalled carbon nanotubes (MWCNTs) is reported. While the frequencies of the tangential modes of carbon nanotubes (G band) harden as the pressure increases, the vibrational frequencies of the chain modes (around 1850 cm–1) decrease, thus indicating a softening of the carbon–carbon bonds in this 1D solid. Pressure-induced irreversible structural changes in the linear carbon chains are unveiled by the red shift in the vibrational modes when pressure is released. These results have been interpreted as being due to a coalescence of carbon chains, and this hypothesis is supported by state-of-the-art atomistic reactive molecular dynamics simulations.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nadia F. Andradea Gustavo Brunettoa, Douglas S. Galvao
High Pressure Induced Binding Between Linear Carbon Chains and Nanotubes Proceedings
vol. 1752, no. 53-58, 2015, (MRS Proceedings, 1752, pp 53-58).
@proceedings{Brunettoa2015,
title = {High Pressure Induced Binding Between Linear Carbon Chains and Nanotubes},
author = {Gustavo Brunettoa, Nadia F. Andradea, Douglas S. Galvao, Antonio G. Souza Filho},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=9553206&fileId=S1946427415000913},
doi = {10.1557/opl.2015.91},
year = {2015},
date = {2015-01-01},
volume = {1752},
number = {53-58},
abstract = {Recent studies of single-walled carbon nanotubes (CNTs) in aqueous media have showed that water can significantly affect the tube mechanical properties. CNTs under hydrostatic compression can preserve their elastic properties up to large pressure values, while exhibiting exceptional resistance to mechanical loadings. It was experimentally observed that CNTs with encapsulated linear carbon chains (LCCs), when subjected to high hydrostatic pressure values, present irreversible red shifts in some of their vibrational frequencies. In order to address the cause of this phenomenon, we have carried out fully atomistic reactive (ReaxFF) molecular dynamics (MD) simulations for model structures mimicking the experimental conditions. We have considered the cases of finite and infinite (cyclic boundary conditions) CNTs filled with LCCs (LCC@CNTs) of different lengths (from 9 up to 40 atoms). Our results show that increasing the hydrostatic pressure causes the CNT to be deformed in an inhomogeneous way due to the LCC presence. The LCC/CNT interface regions exhibit convex curvatures, which results in more reactive sites, thus favoring the formation of covalent chemical bonds between the chain and the nanotube. This process is irreversible with the newly formed bonds continuing to exist even after releasing the external pressure and causing an irreversibly red shift in the chain vibrational modes from 1850 to 1500 cm−1.},
note = {MRS Proceedings, 1752, pp 53-58},
keywords = {},
pubstate = {published},
tppubtype = {proceedings}
}
Vasconcelos, MS; Azevedo, David L; Hadad, A; Galvao, DS
Electronic properties of Fibonacci and random Si--Ge chains Journal Article
In: Journal of Physics: Condensed Matter, vol. 23, no. 40, pp. 405501, 2011.
@article{vasconcelos2011electronic,
title = {Electronic properties of Fibonacci and random Si--Ge chains},
author = {Vasconcelos, MS and Azevedo, David L and Hadad, A and Galvao, DS},
url = {http://iopscience.iop.org/0953-8984/23/40/405501},
year = {2011},
date = {2011-01-01},
journal = {Journal of Physics: Condensed Matter},
volume = {23},
number = {40},
pages = {405501},
publisher = {IOP Publishing},
abstract = {In this paper we address a theoretical calculation of the electronic spectra of an Si–Ge atomic chain that is arranged in a Fibonacci quasi-periodic sequence, by using a semi-empirical quantum method based on the Hückel extended model. We apply the Fibonacci substitutional sequences in the atomic building blocks A(Si) and B(Ge) through the inflation rule or a recursion relation. In our ab initio calculations we use only a single point, which is sufficient for considering all the orbitals and charge distribution across the entire system. Although the calculations presented here are more complete than the models adopted in the literature which take into account the electronic interaction only up to the second and third neighbors, an interesting property remains in their electronic spectra: the fractality (which is the main signature of this kind of system). We discuss this fractality of the spectra and we compare them with the random arrangement of the Si–Ge atomic chain, and with previous results based on the tight-binding approximation of the Schrödinger equation considering up to the nearest neighbor.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Troche, KS; Coluci, VR; Rurali, R; Galvao, DS
Structural and electronic properties of zigzag carbon nanotubes filled with small fullerenes Journal Article
In: Journal of Physics: Condensed Matter, vol. 19, no. 23, pp. 236222, 2007.
@article{troche2007structural,
title = {Structural and electronic properties of zigzag carbon nanotubes filled with small fullerenes},
author = {Troche, KS and Coluci, VR and Rurali, R and Galvao, DS},
url = {http://iopscience.iop.org/0953-8984/19/23/236222},
year = {2007},
date = {2007-01-01},
journal = {Journal of Physics: Condensed Matter},
volume = {19},
number = {23},
pages = {236222},
publisher = {IOP Publishing},
abstract = {In this work we investigated the encapsulation of C20 and C30 fullerenes into semiconducting carbon nanotubes to study the possibility of bandgap engineering in such systems. Classical molecular dynamics simulations coupled to tight-binding calculations were used to determine the conformational and electronic properties of carbon nanotubes with an increasing fullerene concentration. We have observed that C20 fullerenes behave similarly to a n-type dopant while C30 can provide p-type doping in some cases. The combined incorporation of both types of fullerenes (hybrid encapsulation) into the same nanotube leads to a behaviour similar to that found in electronic pn-junctions. These aspects can be exploited in the design of nanoelectronic devices using semiconducting carbon nanotubes.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Sato, Fernando; Braga, Scheila F; Santos, Helio F dos; Galvao, Douglas S
Structure-Activity Relationship Investigation of Some New Tetracyclines by Electronic Index Methodology Journal Article
In: arXiv preprint arXiv:0708.2931, 2007.
@article{sato2007structure,
title = {Structure-Activity Relationship Investigation of Some New Tetracyclines by Electronic Index Methodology},
author = {Sato, Fernando and Braga, Scheila F and Santos, Helio F dos and Galvao, Douglas S},
url = {http://arxiv.org/abs/0708.2931},
year = {2007},
date = {2007-01-01},
journal = {arXiv preprint arXiv:0708.2931},
abstract = {Tetracyclines are an old class of molecules that constitute a broad-spectrum antibiotics. Since the first member of tetracycline family were isolated, the clinical importance of these compounds as therapeutic and prophylactic agents against a wide range of infections has stimulated efforts to define their mode of action as inhibitors of bacterial reproduction. We used three SAR methodologies for the analysis of biological activity of a set of 104 tetracycline compounds. Our calculation were carried out using the semi-empirical Austin Method One (AM1) and Parametric Method 3 (PM3). Electronic Indices Methodology (EIM), Principal Component Analysis (PCA) and Artificial Neural Networks (ANN) were applied to the classification of 14 old and 90 new proposed derivatives of tetracyclines. Our results make evident the importance of EIM descriptors in pattern recognition and also show that the EIM can be effectively used to predict the biological activity of Tetracyclines.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Rurali, R; Coluci, VR; Galvao, DS
Prediction of Giant Electro-actuation for Carbon Nanoscrolls Journal Article
In: arXiv preprint cond-mat/0603239, 2006.
@article{rurali2006predictionb,
title = {Prediction of Giant Electro-actuation for Carbon Nanoscrolls},
author = {Rurali, R and Coluci, VR and Galvao, DS},
url = {http://arxiv.org/abs/cond-mat/0603239},
year = {2006},
date = {2006-01-01},
journal = {arXiv preprint cond-mat/0603239},
abstract = {We study by first-principles calculations the electro-mechanical response of carbon nanoscrolls. We show that although they present a very similar behavior to carbon nanotubes for what concerns the axial deformation sensitivity, they exhibit a radial response upon charge injection which is up to one order of magnitude larger. In association with their high stability, this behavior make them a natural choice for a new class of very efficient nano-actuators.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Rurali, R; Coluci, VR; Galvao, DS
Prediction of giant electroactuation for papyruslike carbon nanoscroll structures: first-principles calculations Journal Article
In: Physical Review B, vol. 74, no. 8, pp. 085414, 2006.
@article{rurali2006prediction,
title = {Prediction of giant electroactuation for papyruslike carbon nanoscroll structures: first-principles calculations},
author = {Rurali, R and Coluci, VR and Galvao, DS},
url = {http://journals.aps.org/prb/abstract/10.1103/PhysRevB.74.085414},
year = {2006},
date = {2006-01-01},
journal = {Physical Review B},
volume = {74},
number = {8},
pages = {085414},
publisher = {American Physical Society},
abstract = {We study by first-principles calculations the electromechanical response of carbon nanoscroll structures. We show that although they present a very similar behavior to carbon nanotubes in their axial deformation sensitivity, they exhibit a radial response upon charge injection which is up to one order of magnitude larger. In association with their high stability, this behavior makes them a natural choice for a new class of very efficient nanoactuators.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Lorite, Gabriela S; Coluci, Vitor R; da Silva, Maria Ivonete N; Deziderio, Shirlei N; Graeff, Carlos Frederico O; Galvao, Douglas S; Cotta, Monica A
Synthetic melanin films: Assembling mechanisms, scaling behavior, and structural properties Journal Article
In: Journal of Applied Physics, vol. 99, no. 11, pp. 113511, 2006.
@article{lorite2006synthetic,
title = {Synthetic melanin films: Assembling mechanisms, scaling behavior, and structural properties},
author = {Lorite, Gabriela S and Coluci, Vitor R and da Silva, Maria Ivonete N and Deziderio, Shirlei N and Graeff, Carlos Frederico O and Galvao, Douglas S and Cotta, Monica A},
url = {http://scitation.aip.org/content/aip/journal/jap/99/11/10.1063/1.2200401},
year = {2006},
date = {2006-01-01},
journal = {Journal of Applied Physics},
volume = {99},
number = {11},
pages = {113511},
publisher = {AIP Publishing},
abstract = {In this work we report on the surface characterization of melanin thin films prepared using both water-based and organic solvent-based melanin syntheses. Atomic force microscopy(AFM) analysis of these films suggests that the organic solvent synthesis provides relatively planar basic melanin structures; these basic structures generate surface steps with height in the range of 2–3nm and small tendency to form larger aggregates. The scaling properties obtained from the AFM data were used to infer the assembling mechanisms of these thin films which depend on the solvent used for melanin synthesis. The behavior observed in organic solvent-based melanin suggests a diffusion-limited aggregation process. Thus films with good adhesion to the substrate and smoother morphologies than water-prepared melanin films are obtained. Electronic structure calculations using a conductorlike screening model were also performed in order to elucidate the microscopic processes of thin film formation. Our results suggest that the agglomerates observed in hydrated samples originate from reaction with water at specific locations on the surface most likely defects on the planar structure.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Giro, Ronaldo; Caldas, Mar'ilia Junqueira; Galvao, Douglas Soares
Band gap engineering for poly (p-phenylene) and poly (p-phenylene vinylene) copolymers using the tight-binding approach Journal Article
In: International Journal of Quantum Chemistry, vol. 103, no. 5, pp. 588–596, 2005.
@article{giro2005band,
title = {Band gap engineering for poly (p-phenylene) and poly (p-phenylene vinylene) copolymers using the tight-binding approach},
author = {Giro, Ronaldo and Caldas, Mar'ilia Junqueira and Galvao, Douglas Soares},
url = {http://onlinelibrary.wiley.com/doi/10.1002/qua.20551/full},
year = {2005},
date = {2005-01-01},
journal = {International Journal of Quantum Chemistry},
volume = {103},
number = {5},
pages = {588--596},
publisher = {Wiley Online Library},
abstract = {The interest in poly(p-phenylene) (PPP) and poly(p-phenylene vinylene) (PPV) copolymers stems from the fact that these homopolymers present interesting optical and electronic properties that allow a great variety of technological applications. Combining different numbers of PPP and PPV units it is possible, in principle, to obtain new structures presenting intermediate gap values (2.8 eV and 2.4 eV for PPP and PPV, respectively). For this study we used a Hückel Hamiltonian tight-binding coupled to the negative factor counting (NFC) technique. We carried out a systematic search to determine optimum relative concentrations for disordered binary polymeric alloys with predefined gap values. Once these structures were obtained, we used the semiempirical methods AM1/PM3 and ZINDO/S-CI for geometrical and optical studies, respectively. Our theoretical results show that it is possible to obtain copolymers of PPP and PPV with intermediate gap values of their parent structures. © 2005 Wiley Periodicals, Inc. Int J Quantum Chem, 2005},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Konstantinova, Elena; Galvao, Douglas S; Barone, Paulo MVB; Dantas, Socrates O
Structural and electronic properties of radialenes and related systems Journal Article
In: Journal of Molecular Structure: THEOCHEM, vol. 729, no. 3, pp. 203–210, 2005.
@article{konstantinova2005structural,
title = {Structural and electronic properties of radialenes and related systems},
author = {Konstantinova, Elena and Galvao, Douglas S and Barone, Paulo MVB and Dantas, Socrates O},
url = {http://www.sciencedirect.com/science/article/pii/S016612800500480X},
year = {2005},
date = {2005-01-01},
journal = {Journal of Molecular Structure: THEOCHEM},
volume = {729},
number = {3},
pages = {203--210},
publisher = {Elsevier},
abstract = {The discovery of new allotropic forms of carbon gives rise to a great interest in carbon compounds as building blocks for novel nanostructure materials. Radialenes are homologous series of compounds with a cycloalkane nucleus bound to methylene side groups, with molecular formula CnHn. The series of expanded radialenes of molecular formulae C2nHn and C3nHn are obtained by inserting acetylene or diacetylene groups between each pair of methylene units. This paper is a report on the theoretical study of structural, electronic and spectroscopic properties of radialenes, expanded radialenes and related molecular systems. Using semiempirical methods we explore the behavior of π-electrons along the carbon-rich skeleton. The results for structural parameters are in a good agreement with the available experimental data. The calculated electronic gaps and spatial distribution of frontier orbitals indicate to interesting electrical and nonlinear optical properties of the explored compounds, which may be useful for technological applications.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Braga, Scheila Furtado; Galvao, Douglas Soares
Benzo [c] quinolizin-3-ones theoretical investigation: SAR analysis and application to nontested compounds Journal Article
In: Journal of chemical information and computer sciences, vol. 44, no. 6, pp. 1987–1997, 2004.
@article{braga2004benzo,
title = {Benzo [c] quinolizin-3-ones theoretical investigation: SAR analysis and application to nontested compounds},
author = {Braga, Scheila Furtado and Galvao, Douglas Soares},
url = {http://pubs.acs.org/doi/abs/10.1021/ci049837u},
year = {2004},
date = {2004-01-01},
journal = {Journal of chemical information and computer sciences},
volume = {44},
number = {6},
pages = {1987--1997},
publisher = {American Chemical Society},
abstract = {We investigate with the use of theoretical methodologies the activity of a set of 41 benzo[c]quinolizin-3-
ones (BC3), some of them explored as selective inhibitors of the human 5R-reductase steroid. For the
structure-activity study we have considered dividing the molecules into groups of tested and nontested
compounds. Semiempirical calculations and pattern recognition methods such as Electronic Indices
Methodology (EIM), Principal Components Analysis (PCA), Hierarchical Cluster Analysis (HCA), and
K-Nearest Neighbors (KNN) have been applied to search for a correlation between experimental activity
and theoretical descriptors. Our results show that it is possible to directly correlate some molecular quantum
descriptors with BC3 biological activity. This information can be used in principle to identify active/inactive
untested compounds and/or to design new active compounds.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
ones (BC3), some of them explored as selective inhibitors of the human 5R-reductase steroid. For the
structure-activity study we have considered dividing the molecules into groups of tested and nontested
compounds. Semiempirical calculations and pattern recognition methods such as Electronic Indices
Methodology (EIM), Principal Components Analysis (PCA), Hierarchical Cluster Analysis (HCA), and
K-Nearest Neighbors (KNN) have been applied to search for a correlation between experimental activity
and theoretical descriptors. Our results show that it is possible to directly correlate some molecular quantum
descriptors with BC3 biological activity. This information can be used in principle to identify active/inactive
untested compounds and/or to design new active compounds.
Vendrame, R; Coluci, Vitor Rafael; Galvao, Douglas Soares
Comparative parametric method 5 (PM5) study of trans-stilbene Journal Article
In: Journal of Molecular Structure: THEOCHEM, vol. 686, no. 1, pp. 103–108, 2004.
@article{vendrame2004comparative,
title = {Comparative parametric method 5 (PM5) study of trans-stilbene},
author = {Vendrame, R and Coluci, Vitor Rafael and Galvao, Douglas Soares},
url = {http://www.sciencedirect.com/science/article/pii/S0166128004005998},
year = {2004},
date = {2004-01-01},
journal = {Journal of Molecular Structure: THEOCHEM},
volume = {686},
number = {1},
pages = {103--108},
publisher = {Elsevier},
abstract = {In this work we report a comparative Austin method 1 (AM1), parametric method 3 (PM3), and parametric method 5 (PM5) studies for trans-stilbene in its ground, excited (singlet and triplet), and ionic (positive and negative polarons and bipolarons) states. We evaluated the accuracy of the recently developed PM5 method. PM5 and AM1 predict a non-planar ground and singlet states for trans-stilbene, while PM3 predicts planar ones, which is in agreement with the available experimental data. In general the PM3 and PM5 bond lengths are superior to AM1 while AM1 bond angles are superior to PM3 and PM5 when compared with available experimental data. The PM5 underestimates the cis–trans isomerization energy and and it is not a quite reliable method for the calculation of relative IP values. The presumed PM5 superior performance against AM1 and PM3 was not observed for the stilbene structures.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Legoas, Sergio B; Rodrigues, Varlei; Ugarte, Daniel; Galvao, Douglas S
Contaminants in suspended gold chains: An ab initio molecular dynamics study Journal Article
In: Physical Review Letters, vol. 93, no. 21, pp. 216103, 2004.
@article{legoas2004contaminants,
title = {Contaminants in suspended gold chains: An ab initio molecular dynamics study},
author = {Legoas, Sergio B and Rodrigues, Varlei and Ugarte, Daniel and Galvao, Douglas S},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.93.216103},
year = {2004},
date = {2004-01-01},
journal = {Physical Review Letters},
volume = {93},
number = {21},
pages = {216103},
publisher = {American Physical Society},
abstract = {Recently, we have proposed that the origin of anomalously long interatomic distances in suspended gold chains could be the result of carbon contamination during sample manipulation [S. B. Legoas et al., Phys. Rev. Lett. 88, 076105 (2002)]. More recently, however, other works have proposed that hydrogen instead of carbon should be the most probable contaminant. We report ab initio molecular dynamics results for different temperatures considering different possible contaminants. Our results show that at nonzero temperatures (more realistic to simulate the experimental conditions) hydrogen may be ruled out and carbon atoms remain the best candidate for contamination.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
dos Santos, Adenilson O; Avanci, Luis H; Cardoso, Lisandro P; Giro, Ronaldo; Legoas, Sergio B; Galvao, Douglas S; Sherwood, John N
Hysteresis-like behavior in MBANP crystals Journal Article
In: Crystal growth & design, vol. 4, no. 5, pp. 1079–1081, 2004.
@article{dos2004hysteresis,
title = {Hysteresis-like behavior in MBANP crystals},
author = {dos Santos, Adenilson O and Avanci, Luis H and Cardoso, Lisandro P and Giro, Ronaldo and Legoas, Sergio B and Galvao, Douglas S and Sherwood, John N},
url = {http://pubs.acs.org/doi/abs/10.1021/cg0341860},
year = {2004},
date = {2004-01-01},
journal = {Crystal growth & design},
volume = {4},
number = {5},
pages = {1079--1081},
publisher = {ACS Publications},
abstract = {he measured strain versus E-cycle in a MBANP organic single crystal showed interesting butterfly wing shape hysteresis behavior. Quantum mechanical calculations on isolated MBANP molecules have shown that the main features of the hysteresis shape can be explained in terms of field induced changes in the charge profiles and geometry of isolated MBANP molecules.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Coluci, VR; Braga, SF; Legoas, SB; Galvao, DS; Baughman, RH
New families of carbon nanotubes based on graphyne motifs Journal Article
In: Nanotechnology, vol. 15, no. 4, pp. S142, 2004.
@article{coluci2004new,
title = {New families of carbon nanotubes based on graphyne motifs},
author = {Coluci, VR and Braga, SF and Legoas, SB and Galvao, DS and Baughman, RH},
url = {http://iopscience.iop.org/0957-4484/15/4/006},
year = {2004},
date = {2004-01-01},
journal = {Nanotechnology},
volume = {15},
number = {4},
pages = {S142},
publisher = {IOP Publishing},
abstract = {Electronic properties of proposed new families of carbon single walled nanotubes are investigated. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Analogous to ordinary nanotubes, armchair, zigzag and chiral graphyne nanotubes are possible. Tight-binding and ab initio density functional methods were used to predict the electronic properties of these unusual nanotubes. Of the three graphyne nanotube families analysed here, two provide metallic behaviour for armchair tubes and either metallic or semiconducting behaviour for zigzag nanotubes. For the other graphyne nanotube family investigated a diameter and chirality independent bandgap is predicted and a bandgap modulation study by structural distortions has been carried out for small longitudinal tube deformations. Interestingly, while the bandgap is insensitive to structure, the stress-induced bandgap changes can strongly depend both on the nanotube type and whether the strain is tensile or compressive.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
De Castro, MPP; Von Zuben, AA; Frateschi, NC; Santo, LLE; Galvao, DS; Bettini, J; De Carvalho, MMG
Strong spatial beryllium doping selectivity on InGaP layers grown on pre-patterned GaAs substrates by chemical beam epitaxy Journal Article
In: Journal of crystal growth, vol. 266, no. 4, pp. 429–434, 2004.
@article{de2004strong,
title = {Strong spatial beryllium doping selectivity on InGaP layers grown on pre-patterned GaAs substrates by chemical beam epitaxy},
author = {De Castro, MPP and Von Zuben, AA and Frateschi, NC and Santo, LLE and Galvao, DS and Bettini, J and De Carvalho, MMG},
url = {http://www.sciencedirect.com/science/article/pii/S0022024804002829},
year = {2004},
date = {2004-01-01},
journal = {Journal of crystal growth},
volume = {266},
number = {4},
pages = {429--434},
publisher = {North-Holland},
abstract = {We present an investigation of beryllium doping selectivity in InGaP layers grown by chemical beam epitaxy on pre-patterned substrates. We observed a resistivity of 3.1×10−2 and 4.5×10−2 Ω cm for (1 1 1)A planes with the growth at 500°C and 540°C, respectively. The layers on (0 0 1) planes show a resistivity of 8.9×10−1 Ω cm with the growth at 500°C, being essentially undoped with the growth at 540°C ⋅ We show how this strong doping selectivity can be explained by Be3P2 cluster formation growth, which depends on growth temperature and planar crystalline structure.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Galvao, Douglas Soares; Rodrigues, Varlei; Ugarte, Daniel; Legoas, Sergio Benites
The role of carbon contamination in metallic nanowires Journal Article
In: Materials Research, vol. 7, no. 2, pp. 339–342, 2004.
@article{galvao2004role,
title = {The role of carbon contamination in metallic nanowires},
author = {Galvao, Douglas Soares and Rodrigues, Varlei and Ugarte, Daniel and Legoas, Sergio Benites},
url = {http://www.scielo.br/scielo.php?pid=S1516-14392004000200020&script=sci_arttext},
year = {2004},
date = {2004-01-01},
journal = {Materials Research},
volume = {7},
number = {2},
pages = {339--342},
publisher = {SciELO Brasil},
abstract = {Metallic nanowires have attracted much attention in the last years due to new phenomena such as quantum conductance and the existence of unexpected long interatomic distances attaining 0.3-0.5 nm. These large distances represented a challenge for physical interpretation. In this work we present experimental data from high-resolution transmission electron microscopy and results from ab initio calculations for suspended gold chains and show that these large distances can be easily explained by the presence of carbon atoms as contaminants. In principle the present conclusions can be also applied to other metallic nanowires (such as Ag and Pt) whose structures also present large interatomic distances.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2015
Chandra Sekhar Tiwary Dibyendu Chakravarty, Leonardo Dantas Machado
Zirconia-Nanoparticle-Reinforced Morphology-Engineered Graphene-Based Foams Journal Article
In: Advanced Materials, vol. 27, no. 31, pp. 4534–4543, 2015.
Abstract | Links | BibTeX | Tags: Electronic Structure, Mechanical Properties, Mole, Molecular Dynamics, Nanoparticles, Zirconia
@article{Chakravarty2015,
title = {Zirconia-Nanoparticle-Reinforced Morphology-Engineered Graphene-Based Foams},
author = { Dibyendu Chakravarty , Chandra Sekhar Tiwary , Leonardo Dantas Machado ,
Gustavo Brunetto , Soumya Vinod , Ram Manohar Yadav , Douglas S. Galvao ,
Shrikant V. Joshi , Govindan Sundararajan, Pulickel M. Ajayan },
url = {http://onlinelibrary.wiley.com/doi/10.1002/adma.201502409/full},
doi = {10.1002/adma.201502409},
year = {2015},
date = {2015-07-15},
journal = {Advanced Materials},
volume = {27},
number = {31},
pages = {4534–4543},
abstract = {The morphology of graphene-based foams can be engineered by reinforcing them with nanocrystalline zirconia, thus improving their oil-adsorption capacity; This can be observed experimentally and explained theoretically. Low zirconia fractions yield flaky microstructures where zirconia nanoparticles arrest propagating cracks. Higher zirconia concentrations possess a mesh-like interconnected structure where the degree of coiling is dependant on the local zirconia content.},
keywords = {Electronic Structure, Mechanical Properties, Mole, Molecular Dynamics, Nanoparticles, Zirconia},
pubstate = {published},
tppubtype = {article}
}
Yongji Gong Kunttal Keyshar, Gonglan Ye
Chemical Vapor Deposition of Monolayer Rhenium Disulfide (ReS2) Journal Article
In: Advanced Materials, vol. 27, no. 31, pp. 4640–4648, 2015.
Abstract | Links | BibTeX | Tags: Electronic Structure, Rhenium Disulfide, Synthesis
@article{Keyshar2015,
title = {Chemical Vapor Deposition of Monolayer Rhenium Disulfide (ReS2)},
author = {Kunttal Keyshar , Yongji Gong , Gonglan Ye , Gustavo Brunetto , Wu Zhou ,
Daniel P. Cole , Ken Hackenberg , Yongmin He , Leonardo Machado , Mohamad Kabbani ,
Amelia H. C. Hart , Bo Li , Douglas S. Galvao , Antony George , Robert Vajtai ,
Chandra Sekhar Tiwary , Pulickel M. Ajayan},
url = {http://onlinelibrary.wiley.com/doi/10.1002/adma.201501795/full},
doi = {10.1002/adma.201501795},
year = {2015},
date = {2015-07-03},
journal = {Advanced Materials},
volume = {27},
number = {31},
pages = {4640–4648},
abstract = {The direct synthesis of monolayer and multilayer ReS2 by chemical vapor deposition at a low temperature of 450 °C is reported. Detailed characterization of this material is performed using various spectroscopy and microscopy methods. Furthermore initial field-effect transistor characteristics are evaluated, which highlight the potential in being used as an n-type semiconductor.},
keywords = {Electronic Structure, Rhenium Disulfide, Synthesis},
pubstate = {published},
tppubtype = {article}
}
Chandra Sekhar Tiwary Mohamad A Kabbani, Pedro AS Autreto
Ambient solid-state mechano-chemical reactions between functionalized carbon nanotubes Journal Article
In: Nature Communications, vol. 6, pp. 7291, 2015.
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, Chemical Reactions, Electronic Structure, Molecular Dynamics, top20
@article{Kabbani2015,
title = {Ambient solid-state mechano-chemical reactions between functionalized carbon nanotubes},
author = {Mohamad A Kabbani, Chandra Sekhar Tiwary, Pedro AS Autreto, Gustavo Brunetto, Anirban Som, KR Krishnadas, Sehmus Ozden, Ken P Hackenberg, Yongi Gong, Douglas S Galvao, Robert Vajtai, Ahmad T Kabbani, Thalappil Pradeep, Pulickel M Ajayan},
url = {http://www.nature.com/ncomms/2015/150615/ncomms8291/full/ncomms8291.html},
doi = {10.1038/ncomms8291},
year = {2015},
date = {2015-06-15},
journal = {Nature Communications},
volume = {6},
pages = {7291},
abstract = {Carbon nanotubes can be chemically modified by attaching various functionalities to their surfaces, although harsh chemical treatments can lead to their break-up into graphene nanostructures. On the other hand, direct coupling between functionalities bound on individual nanotubes could lead to, as yet unexplored, spontaneous chemical reactions. Here we report an ambient mechano-chemical reaction between two varieties of nanotubes, carrying predominantly carboxyl and hydroxyl functionalities, respectively, facilitated by simple mechanical grinding of the reactants. The purely solid-state reaction between the chemically differentiated nanotube species produces condensation products and unzipping of nanotubes due to local energy release, as confirmed by spectroscopic measurements, thermal analysis and molecular dynamic simulations.},
keywords = {Carbon Nanotubes, Chemical Reactions, Electronic Structure, Molecular Dynamics, top20},
pubstate = {published},
tppubtype = {article}
}
Acrísio L Aguiar Nadia Ferreira Andrade, Yoong Ahm Kim
Linear Carbon Chains Under High Pressure Conditions Journal Article
In: The Journal of Physical Chemistry C, vol. 119, no. 19, pp. 10669–10676, 2015.
Abstract | Links | BibTeX | Tags: Atomic Chains, Carbon Nanotubes, Electronic Structure, Molecular Dynamics, Raman
@article{Andrade2015,
title = {Linear Carbon Chains Under High Pressure Conditions},
author = {Nadia Ferreira Andrade, Acrísio L Aguiar, Yoong Ahm Kim, Morinobu Endo, Paulo TC Freire, Gustavo Bruneto, Douglas Soares Galvao, Mildred S Dresselhaus, Antonio Gomes Souza Filho},
url = {http://pubs.acs.org/doi/abs/10.1021/acs.jpcc.5b00902},
doi = {10.1021/acs.jpcc.5b00902},
year = {2015},
date = {2015-04-23},
journal = {The Journal of Physical Chemistry C},
volume = {119},
number = {19},
pages = {10669–10676},
abstract = {A high-pressure resonance Raman spectroscopy study of linear carbon chains encapsulated inside multiwalled carbon nanotubes (MWCNTs) is reported. While the frequencies of the tangential modes of carbon nanotubes (G band) harden as the pressure increases, the vibrational frequencies of the chain modes (around 1850 cm–1) decrease, thus indicating a softening of the carbon–carbon bonds in this 1D solid. Pressure-induced irreversible structural changes in the linear carbon chains are unveiled by the red shift in the vibrational modes when pressure is released. These results have been interpreted as being due to a coalescence of carbon chains, and this hypothesis is supported by state-of-the-art atomistic reactive molecular dynamics simulations.},
keywords = {Atomic Chains, Carbon Nanotubes, Electronic Structure, Molecular Dynamics, Raman},
pubstate = {published},
tppubtype = {article}
}

Nadia F. Andradea Gustavo Brunettoa, Douglas S. Galvao
High Pressure Induced Binding Between Linear Carbon Chains and Nanotubes Proceedings
vol. 1752, no. 53-58, 2015, (MRS Proceedings, 1752, pp 53-58).
Abstract | Links | BibTeX | Tags: CNT encapsulation, Electronic Structure, Linear Chains, Molecular Dynamics
@proceedings{Brunettoa2015,
title = {High Pressure Induced Binding Between Linear Carbon Chains and Nanotubes},
author = {Gustavo Brunettoa, Nadia F. Andradea, Douglas S. Galvao, Antonio G. Souza Filho},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=9553206&fileId=S1946427415000913},
doi = {10.1557/opl.2015.91},
year = {2015},
date = {2015-01-01},
volume = {1752},
number = {53-58},
abstract = {Recent studies of single-walled carbon nanotubes (CNTs) in aqueous media have showed that water can significantly affect the tube mechanical properties. CNTs under hydrostatic compression can preserve their elastic properties up to large pressure values, while exhibiting exceptional resistance to mechanical loadings. It was experimentally observed that CNTs with encapsulated linear carbon chains (LCCs), when subjected to high hydrostatic pressure values, present irreversible red shifts in some of their vibrational frequencies. In order to address the cause of this phenomenon, we have carried out fully atomistic reactive (ReaxFF) molecular dynamics (MD) simulations for model structures mimicking the experimental conditions. We have considered the cases of finite and infinite (cyclic boundary conditions) CNTs filled with LCCs (LCC@CNTs) of different lengths (from 9 up to 40 atoms). Our results show that increasing the hydrostatic pressure causes the CNT to be deformed in an inhomogeneous way due to the LCC presence. The LCC/CNT interface regions exhibit convex curvatures, which results in more reactive sites, thus favoring the formation of covalent chemical bonds between the chain and the nanotube. This process is irreversible with the newly formed bonds continuing to exist even after releasing the external pressure and causing an irreversibly red shift in the chain vibrational modes from 1850 to 1500 cm−1.},
note = {MRS Proceedings, 1752, pp 53-58},
keywords = {CNT encapsulation, Electronic Structure, Linear Chains, Molecular Dynamics},
pubstate = {published},
tppubtype = {proceedings}
}
2011
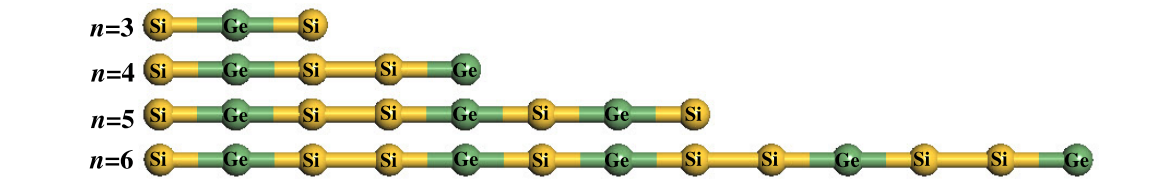
Vasconcelos, MS; Azevedo, David L; Hadad, A; Galvao, DS
Electronic properties of Fibonacci and random Si--Ge chains Journal Article
In: Journal of Physics: Condensed Matter, vol. 23, no. 40, pp. 405501, 2011.
Abstract | Links | BibTeX | Tags: Electronic Structure, Fibonacci, Si-Ge chains
@article{vasconcelos2011electronic,
title = {Electronic properties of Fibonacci and random Si--Ge chains},
author = {Vasconcelos, MS and Azevedo, David L and Hadad, A and Galvao, DS},
url = {http://iopscience.iop.org/0953-8984/23/40/405501},
year = {2011},
date = {2011-01-01},
journal = {Journal of Physics: Condensed Matter},
volume = {23},
number = {40},
pages = {405501},
publisher = {IOP Publishing},
abstract = {In this paper we address a theoretical calculation of the electronic spectra of an Si–Ge atomic chain that is arranged in a Fibonacci quasi-periodic sequence, by using a semi-empirical quantum method based on the Hückel extended model. We apply the Fibonacci substitutional sequences in the atomic building blocks A(Si) and B(Ge) through the inflation rule or a recursion relation. In our ab initio calculations we use only a single point, which is sufficient for considering all the orbitals and charge distribution across the entire system. Although the calculations presented here are more complete than the models adopted in the literature which take into account the electronic interaction only up to the second and third neighbors, an interesting property remains in their electronic spectra: the fractality (which is the main signature of this kind of system). We discuss this fractality of the spectra and we compare them with the random arrangement of the Si–Ge atomic chain, and with previous results based on the tight-binding approximation of the Schrödinger equation considering up to the nearest neighbor.
},
keywords = {Electronic Structure, Fibonacci, Si-Ge chains},
pubstate = {published},
tppubtype = {article}
}
2007
Troche, KS; Coluci, VR; Rurali, R; Galvao, DS
Structural and electronic properties of zigzag carbon nanotubes filled with small fullerenes Journal Article
In: Journal of Physics: Condensed Matter, vol. 19, no. 23, pp. 236222, 2007.
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, CNT encapsulation, Electronic Structure, Fullerenes, Peapods
@article{troche2007structural,
title = {Structural and electronic properties of zigzag carbon nanotubes filled with small fullerenes},
author = {Troche, KS and Coluci, VR and Rurali, R and Galvao, DS},
url = {http://iopscience.iop.org/0953-8984/19/23/236222},
year = {2007},
date = {2007-01-01},
journal = {Journal of Physics: Condensed Matter},
volume = {19},
number = {23},
pages = {236222},
publisher = {IOP Publishing},
abstract = {In this work we investigated the encapsulation of C20 and C30 fullerenes into semiconducting carbon nanotubes to study the possibility of bandgap engineering in such systems. Classical molecular dynamics simulations coupled to tight-binding calculations were used to determine the conformational and electronic properties of carbon nanotubes with an increasing fullerene concentration. We have observed that C20 fullerenes behave similarly to a n-type dopant while C30 can provide p-type doping in some cases. The combined incorporation of both types of fullerenes (hybrid encapsulation) into the same nanotube leads to a behaviour similar to that found in electronic pn-junctions. These aspects can be exploited in the design of nanoelectronic devices using semiconducting carbon nanotubes.
},
keywords = {Carbon Nanotubes, CNT encapsulation, Electronic Structure, Fullerenes, Peapods},
pubstate = {published},
tppubtype = {article}
}
Sato, Fernando; Braga, Scheila F; Santos, Helio F dos; Galvao, Douglas S
Structure-Activity Relationship Investigation of Some New Tetracyclines by Electronic Index Methodology Journal Article
In: arXiv preprint arXiv:0708.2931, 2007.
Abstract | Links | BibTeX | Tags: Drug Design, Electronic Structure, Neural Networks, PCA/HCA, Tetracyclines, Theory of Electronic Indices
@article{sato2007structure,
title = {Structure-Activity Relationship Investigation of Some New Tetracyclines by Electronic Index Methodology},
author = {Sato, Fernando and Braga, Scheila F and Santos, Helio F dos and Galvao, Douglas S},
url = {http://arxiv.org/abs/0708.2931},
year = {2007},
date = {2007-01-01},
journal = {arXiv preprint arXiv:0708.2931},
abstract = {Tetracyclines are an old class of molecules that constitute a broad-spectrum antibiotics. Since the first member of tetracycline family were isolated, the clinical importance of these compounds as therapeutic and prophylactic agents against a wide range of infections has stimulated efforts to define their mode of action as inhibitors of bacterial reproduction. We used three SAR methodologies for the analysis of biological activity of a set of 104 tetracycline compounds. Our calculation were carried out using the semi-empirical Austin Method One (AM1) and Parametric Method 3 (PM3). Electronic Indices Methodology (EIM), Principal Component Analysis (PCA) and Artificial Neural Networks (ANN) were applied to the classification of 14 old and 90 new proposed derivatives of tetracyclines. Our results make evident the importance of EIM descriptors in pattern recognition and also show that the EIM can be effectively used to predict the biological activity of Tetracyclines.},
keywords = {Drug Design, Electronic Structure, Neural Networks, PCA/HCA, Tetracyclines, Theory of Electronic Indices},
pubstate = {published},
tppubtype = {article}
}
2006

Rurali, R; Coluci, VR; Galvao, DS
Prediction of Giant Electro-actuation for Carbon Nanoscrolls Journal Article
In: arXiv preprint cond-mat/0603239, 2006.
Abstract | Links | BibTeX | Tags: DFT, Electroactuation, Electronic Structure, Scrolls
@article{rurali2006predictionb,
title = {Prediction of Giant Electro-actuation for Carbon Nanoscrolls},
author = {Rurali, R and Coluci, VR and Galvao, DS},
url = {http://arxiv.org/abs/cond-mat/0603239},
year = {2006},
date = {2006-01-01},
journal = {arXiv preprint cond-mat/0603239},
abstract = {We study by first-principles calculations the electro-mechanical response of carbon nanoscrolls. We show that although they present a very similar behavior to carbon nanotubes for what concerns the axial deformation sensitivity, they exhibit a radial response upon charge injection which is up to one order of magnitude larger. In association with their high stability, this behavior make them a natural choice for a new class of very efficient nano-actuators.},
keywords = {DFT, Electroactuation, Electronic Structure, Scrolls},
pubstate = {published},
tppubtype = {article}
}
Rurali, R; Coluci, VR; Galvao, DS
Prediction of giant electroactuation for papyruslike carbon nanoscroll structures: first-principles calculations Journal Article
In: Physical Review B, vol. 74, no. 8, pp. 085414, 2006.
Abstract | Links | BibTeX | Tags: DFT, Electronic Structure, Eletroactuation, Scrolls
@article{rurali2006prediction,
title = {Prediction of giant electroactuation for papyruslike carbon nanoscroll structures: first-principles calculations},
author = {Rurali, R and Coluci, VR and Galvao, DS},
url = {http://journals.aps.org/prb/abstract/10.1103/PhysRevB.74.085414},
year = {2006},
date = {2006-01-01},
journal = {Physical Review B},
volume = {74},
number = {8},
pages = {085414},
publisher = {American Physical Society},
abstract = {We study by first-principles calculations the electromechanical response of carbon nanoscroll structures. We show that although they present a very similar behavior to carbon nanotubes in their axial deformation sensitivity, they exhibit a radial response upon charge injection which is up to one order of magnitude larger. In association with their high stability, this behavior makes them a natural choice for a new class of very efficient nanoactuators.},
keywords = {DFT, Electronic Structure, Eletroactuation, Scrolls},
pubstate = {published},
tppubtype = {article}
}
Lorite, Gabriela S; Coluci, Vitor R; da Silva, Maria Ivonete N; Deziderio, Shirlei N; Graeff, Carlos Frederico O; Galvao, Douglas S; Cotta, Monica A
Synthetic melanin films: Assembling mechanisms, scaling behavior, and structural properties Journal Article
In: Journal of Applied Physics, vol. 99, no. 11, pp. 113511, 2006.
Abstract | Links | BibTeX | Tags: Electronic Structure, Melanin, Structure
@article{lorite2006synthetic,
title = {Synthetic melanin films: Assembling mechanisms, scaling behavior, and structural properties},
author = {Lorite, Gabriela S and Coluci, Vitor R and da Silva, Maria Ivonete N and Deziderio, Shirlei N and Graeff, Carlos Frederico O and Galvao, Douglas S and Cotta, Monica A},
url = {http://scitation.aip.org/content/aip/journal/jap/99/11/10.1063/1.2200401},
year = {2006},
date = {2006-01-01},
journal = {Journal of Applied Physics},
volume = {99},
number = {11},
pages = {113511},
publisher = {AIP Publishing},
abstract = {In this work we report on the surface characterization of melanin thin films prepared using both water-based and organic solvent-based melanin syntheses. Atomic force microscopy(AFM) analysis of these films suggests that the organic solvent synthesis provides relatively planar basic melanin structures; these basic structures generate surface steps with height in the range of 2–3nm and small tendency to form larger aggregates. The scaling properties obtained from the AFM data were used to infer the assembling mechanisms of these thin films which depend on the solvent used for melanin synthesis. The behavior observed in organic solvent-based melanin suggests a diffusion-limited aggregation process. Thus films with good adhesion to the substrate and smoother morphologies than water-prepared melanin films are obtained. Electronic structure calculations using a conductorlike screening model were also performed in order to elucidate the microscopic processes of thin film formation. Our results suggest that the agglomerates observed in hydrated samples originate from reaction with water at specific locations on the surface most likely defects on the planar structure.},
keywords = {Electronic Structure, Melanin, Structure},
pubstate = {published},
tppubtype = {article}
}
2005
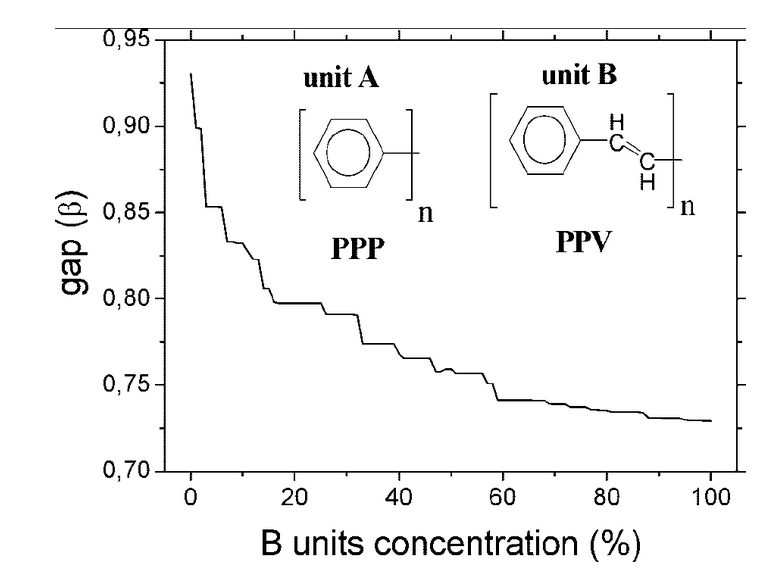
Giro, Ronaldo; Caldas, Mar'ilia Junqueira; Galvao, Douglas Soares
Band gap engineering for poly (p-phenylene) and poly (p-phenylene vinylene) copolymers using the tight-binding approach Journal Article
In: International Journal of Quantum Chemistry, vol. 103, no. 5, pp. 588–596, 2005.
Abstract | Links | BibTeX | Tags: Conducting Polymers, Electronic Structure, PPP, PPV
@article{giro2005band,
title = {Band gap engineering for poly (p-phenylene) and poly (p-phenylene vinylene) copolymers using the tight-binding approach},
author = {Giro, Ronaldo and Caldas, Mar'ilia Junqueira and Galvao, Douglas Soares},
url = {http://onlinelibrary.wiley.com/doi/10.1002/qua.20551/full},
year = {2005},
date = {2005-01-01},
journal = {International Journal of Quantum Chemistry},
volume = {103},
number = {5},
pages = {588--596},
publisher = {Wiley Online Library},
abstract = {The interest in poly(p-phenylene) (PPP) and poly(p-phenylene vinylene) (PPV) copolymers stems from the fact that these homopolymers present interesting optical and electronic properties that allow a great variety of technological applications. Combining different numbers of PPP and PPV units it is possible, in principle, to obtain new structures presenting intermediate gap values (2.8 eV and 2.4 eV for PPP and PPV, respectively). For this study we used a Hückel Hamiltonian tight-binding coupled to the negative factor counting (NFC) technique. We carried out a systematic search to determine optimum relative concentrations for disordered binary polymeric alloys with predefined gap values. Once these structures were obtained, we used the semiempirical methods AM1/PM3 and ZINDO/S-CI for geometrical and optical studies, respectively. Our theoretical results show that it is possible to obtain copolymers of PPP and PPV with intermediate gap values of their parent structures. © 2005 Wiley Periodicals, Inc. Int J Quantum Chem, 2005},
keywords = {Conducting Polymers, Electronic Structure, PPP, PPV},
pubstate = {published},
tppubtype = {article}
}
Konstantinova, Elena; Galvao, Douglas S; Barone, Paulo MVB; Dantas, Socrates O
Structural and electronic properties of radialenes and related systems Journal Article
In: Journal of Molecular Structure: THEOCHEM, vol. 729, no. 3, pp. 203–210, 2005.
Abstract | Links | BibTeX | Tags: DFT, Electronic Structure, Radialenes
@article{konstantinova2005structural,
title = {Structural and electronic properties of radialenes and related systems},
author = {Konstantinova, Elena and Galvao, Douglas S and Barone, Paulo MVB and Dantas, Socrates O},
url = {http://www.sciencedirect.com/science/article/pii/S016612800500480X},
year = {2005},
date = {2005-01-01},
journal = {Journal of Molecular Structure: THEOCHEM},
volume = {729},
number = {3},
pages = {203--210},
publisher = {Elsevier},
abstract = {The discovery of new allotropic forms of carbon gives rise to a great interest in carbon compounds as building blocks for novel nanostructure materials. Radialenes are homologous series of compounds with a cycloalkane nucleus bound to methylene side groups, with molecular formula CnHn. The series of expanded radialenes of molecular formulae C2nHn and C3nHn are obtained by inserting acetylene or diacetylene groups between each pair of methylene units. This paper is a report on the theoretical study of structural, electronic and spectroscopic properties of radialenes, expanded radialenes and related molecular systems. Using semiempirical methods we explore the behavior of π-electrons along the carbon-rich skeleton. The results for structural parameters are in a good agreement with the available experimental data. The calculated electronic gaps and spatial distribution of frontier orbitals indicate to interesting electrical and nonlinear optical properties of the explored compounds, which may be useful for technological applications.},
keywords = {DFT, Electronic Structure, Radialenes},
pubstate = {published},
tppubtype = {article}
}
2004
![Benzo [c] quinolizin-3-ones theoretical investigation: SAR analysis and application to nontested compounds](https://sites.ifi.unicamp.br/galvao/files/2015/02/Screen-Shot-2015-02-26-at-12.21.40-PM.png)
Braga, Scheila Furtado; Galvao, Douglas Soares
Benzo [c] quinolizin-3-ones theoretical investigation: SAR analysis and application to nontested compounds Journal Article
In: Journal of chemical information and computer sciences, vol. 44, no. 6, pp. 1987–1997, 2004.
Abstract | Links | BibTeX | Tags: Drug Design, Electronic Structure, PCA/HCA, Theory of Electronic Indices
@article{braga2004benzo,
title = {Benzo [c] quinolizin-3-ones theoretical investigation: SAR analysis and application to nontested compounds},
author = {Braga, Scheila Furtado and Galvao, Douglas Soares},
url = {http://pubs.acs.org/doi/abs/10.1021/ci049837u},
year = {2004},
date = {2004-01-01},
journal = {Journal of chemical information and computer sciences},
volume = {44},
number = {6},
pages = {1987--1997},
publisher = {American Chemical Society},
abstract = {We investigate with the use of theoretical methodologies the activity of a set of 41 benzo[c]quinolizin-3-
ones (BC3), some of them explored as selective inhibitors of the human 5R-reductase steroid. For the
structure-activity study we have considered dividing the molecules into groups of tested and nontested
compounds. Semiempirical calculations and pattern recognition methods such as Electronic Indices
Methodology (EIM), Principal Components Analysis (PCA), Hierarchical Cluster Analysis (HCA), and
K-Nearest Neighbors (KNN) have been applied to search for a correlation between experimental activity
and theoretical descriptors. Our results show that it is possible to directly correlate some molecular quantum
descriptors with BC3 biological activity. This information can be used in principle to identify active/inactive
untested compounds and/or to design new active compounds.},
keywords = {Drug Design, Electronic Structure, PCA/HCA, Theory of Electronic Indices},
pubstate = {published},
tppubtype = {article}
}
ones (BC3), some of them explored as selective inhibitors of the human 5R-reductase steroid. For the
structure-activity study we have considered dividing the molecules into groups of tested and nontested
compounds. Semiempirical calculations and pattern recognition methods such as Electronic Indices
Methodology (EIM), Principal Components Analysis (PCA), Hierarchical Cluster Analysis (HCA), and
K-Nearest Neighbors (KNN) have been applied to search for a correlation between experimental activity
and theoretical descriptors. Our results show that it is possible to directly correlate some molecular quantum
descriptors with BC3 biological activity. This information can be used in principle to identify active/inactive
untested compounds and/or to design new active compounds.
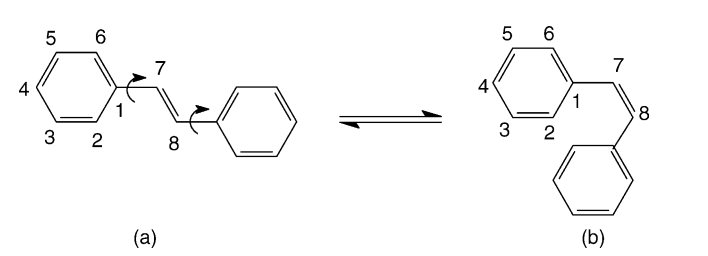
Vendrame, R; Coluci, Vitor Rafael; Galvao, Douglas Soares
Comparative parametric method 5 (PM5) study of trans-stilbene Journal Article
In: Journal of Molecular Structure: THEOCHEM, vol. 686, no. 1, pp. 103–108, 2004.
Abstract | Links | BibTeX | Tags: Electronic Structure, MOPAC, PM3, PM5, Stilbene
@article{vendrame2004comparative,
title = {Comparative parametric method 5 (PM5) study of trans-stilbene},
author = {Vendrame, R and Coluci, Vitor Rafael and Galvao, Douglas Soares},
url = {http://www.sciencedirect.com/science/article/pii/S0166128004005998},
year = {2004},
date = {2004-01-01},
journal = {Journal of Molecular Structure: THEOCHEM},
volume = {686},
number = {1},
pages = {103--108},
publisher = {Elsevier},
abstract = {In this work we report a comparative Austin method 1 (AM1), parametric method 3 (PM3), and parametric method 5 (PM5) studies for trans-stilbene in its ground, excited (singlet and triplet), and ionic (positive and negative polarons and bipolarons) states. We evaluated the accuracy of the recently developed PM5 method. PM5 and AM1 predict a non-planar ground and singlet states for trans-stilbene, while PM3 predicts planar ones, which is in agreement with the available experimental data. In general the PM3 and PM5 bond lengths are superior to AM1 while AM1 bond angles are superior to PM3 and PM5 when compared with available experimental data. The PM5 underestimates the cis–trans isomerization energy and and it is not a quite reliable method for the calculation of relative IP values. The presumed PM5 superior performance against AM1 and PM3 was not observed for the stilbene structures.
},
keywords = {Electronic Structure, MOPAC, PM3, PM5, Stilbene},
pubstate = {published},
tppubtype = {article}
}
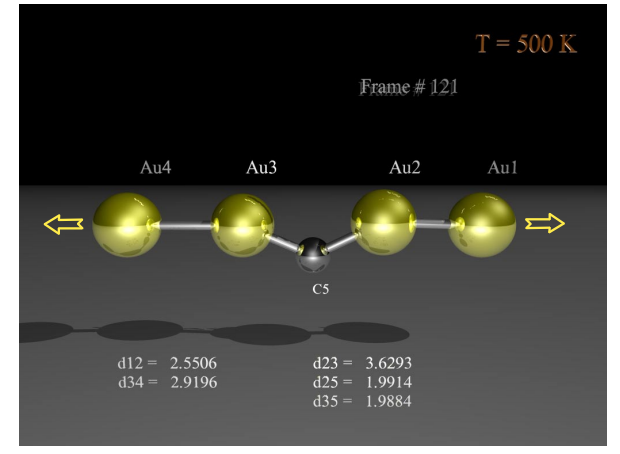
Legoas, Sergio B; Rodrigues, Varlei; Ugarte, Daniel; Galvao, Douglas S
Contaminants in suspended gold chains: An ab initio molecular dynamics study Journal Article
In: Physical Review Letters, vol. 93, no. 21, pp. 216103, 2004.
Abstract | Links | BibTeX | Tags: DFT, Electronic Structure, Linear Atomic Chains, Metallic Nanowires, top20
@article{legoas2004contaminants,
title = {Contaminants in suspended gold chains: An ab initio molecular dynamics study},
author = {Legoas, Sergio B and Rodrigues, Varlei and Ugarte, Daniel and Galvao, Douglas S},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.93.216103},
year = {2004},
date = {2004-01-01},
journal = {Physical Review Letters},
volume = {93},
number = {21},
pages = {216103},
publisher = {American Physical Society},
abstract = {Recently, we have proposed that the origin of anomalously long interatomic distances in suspended gold chains could be the result of carbon contamination during sample manipulation [S. B. Legoas et al., Phys. Rev. Lett. 88, 076105 (2002)]. More recently, however, other works have proposed that hydrogen instead of carbon should be the most probable contaminant. We report ab initio molecular dynamics results for different temperatures considering different possible contaminants. Our results show that at nonzero temperatures (more realistic to simulate the experimental conditions) hydrogen may be ruled out and carbon atoms remain the best candidate for contamination.},
keywords = {DFT, Electronic Structure, Linear Atomic Chains, Metallic Nanowires, top20},
pubstate = {published},
tppubtype = {article}
}
dos Santos, Adenilson O; Avanci, Luis H; Cardoso, Lisandro P; Giro, Ronaldo; Legoas, Sergio B; Galvao, Douglas S; Sherwood, John N
Hysteresis-like behavior in MBANP crystals Journal Article
In: Crystal growth & design, vol. 4, no. 5, pp. 1079–1081, 2004.
Abstract | Links | BibTeX | Tags: Crystallographic Structure, Electronic Structure, Molecular Crystals
@article{dos2004hysteresis,
title = {Hysteresis-like behavior in MBANP crystals},
author = {dos Santos, Adenilson O and Avanci, Luis H and Cardoso, Lisandro P and Giro, Ronaldo and Legoas, Sergio B and Galvao, Douglas S and Sherwood, John N},
url = {http://pubs.acs.org/doi/abs/10.1021/cg0341860},
year = {2004},
date = {2004-01-01},
journal = {Crystal growth & design},
volume = {4},
number = {5},
pages = {1079--1081},
publisher = {ACS Publications},
abstract = {he measured strain versus E-cycle in a MBANP organic single crystal showed interesting butterfly wing shape hysteresis behavior. Quantum mechanical calculations on isolated MBANP molecules have shown that the main features of the hysteresis shape can be explained in terms of field induced changes in the charge profiles and geometry of isolated MBANP molecules.},
keywords = {Crystallographic Structure, Electronic Structure, Molecular Crystals},
pubstate = {published},
tppubtype = {article}
}
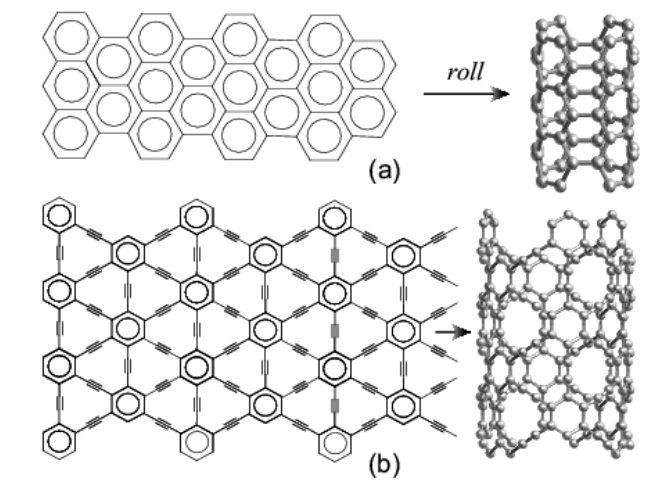
Coluci, VR; Braga, SF; Legoas, SB; Galvao, DS; Baughman, RH
New families of carbon nanotubes based on graphyne motifs Journal Article
In: Nanotechnology, vol. 15, no. 4, pp. S142, 2004.
Abstract | Links | BibTeX | Tags: Allotropes, Electronic Structure, Graphynes, Nanotubes
@article{coluci2004new,
title = {New families of carbon nanotubes based on graphyne motifs},
author = {Coluci, VR and Braga, SF and Legoas, SB and Galvao, DS and Baughman, RH},
url = {http://iopscience.iop.org/0957-4484/15/4/006},
year = {2004},
date = {2004-01-01},
journal = {Nanotechnology},
volume = {15},
number = {4},
pages = {S142},
publisher = {IOP Publishing},
abstract = {Electronic properties of proposed new families of carbon single walled nanotubes are investigated. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Analogous to ordinary nanotubes, armchair, zigzag and chiral graphyne nanotubes are possible. Tight-binding and ab initio density functional methods were used to predict the electronic properties of these unusual nanotubes. Of the three graphyne nanotube families analysed here, two provide metallic behaviour for armchair tubes and either metallic or semiconducting behaviour for zigzag nanotubes. For the other graphyne nanotube family investigated a diameter and chirality independent bandgap is predicted and a bandgap modulation study by structural distortions has been carried out for small longitudinal tube deformations. Interestingly, while the bandgap is insensitive to structure, the stress-induced bandgap changes can strongly depend both on the nanotube type and whether the strain is tensile or compressive.
},
keywords = {Allotropes, Electronic Structure, Graphynes, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
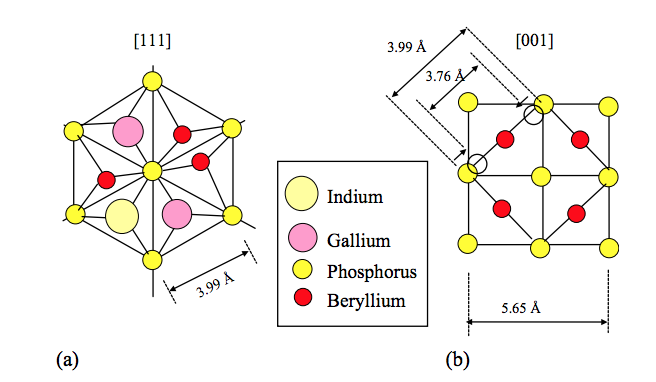
De Castro, MPP; Von Zuben, AA; Frateschi, NC; Santo, LLE; Galvao, DS; Bettini, J; De Carvalho, MMG
Strong spatial beryllium doping selectivity on InGaP layers grown on pre-patterned GaAs substrates by chemical beam epitaxy Journal Article
In: Journal of crystal growth, vol. 266, no. 4, pp. 429–434, 2004.
Abstract | Links | BibTeX | Tags: Crystal Growth, Doping, Electronic Structure
@article{de2004strong,
title = {Strong spatial beryllium doping selectivity on InGaP layers grown on pre-patterned GaAs substrates by chemical beam epitaxy},
author = {De Castro, MPP and Von Zuben, AA and Frateschi, NC and Santo, LLE and Galvao, DS and Bettini, J and De Carvalho, MMG},
url = {http://www.sciencedirect.com/science/article/pii/S0022024804002829},
year = {2004},
date = {2004-01-01},
journal = {Journal of crystal growth},
volume = {266},
number = {4},
pages = {429--434},
publisher = {North-Holland},
abstract = {We present an investigation of beryllium doping selectivity in InGaP layers grown by chemical beam epitaxy on pre-patterned substrates. We observed a resistivity of 3.1×10−2 and 4.5×10−2 Ω cm for (1 1 1)A planes with the growth at 500°C and 540°C, respectively. The layers on (0 0 1) planes show a resistivity of 8.9×10−1 Ω cm with the growth at 500°C, being essentially undoped with the growth at 540°C ⋅ We show how this strong doping selectivity can be explained by Be3P2 cluster formation growth, which depends on growth temperature and planar crystalline structure.},
keywords = {Crystal Growth, Doping, Electronic Structure},
pubstate = {published},
tppubtype = {article}
}
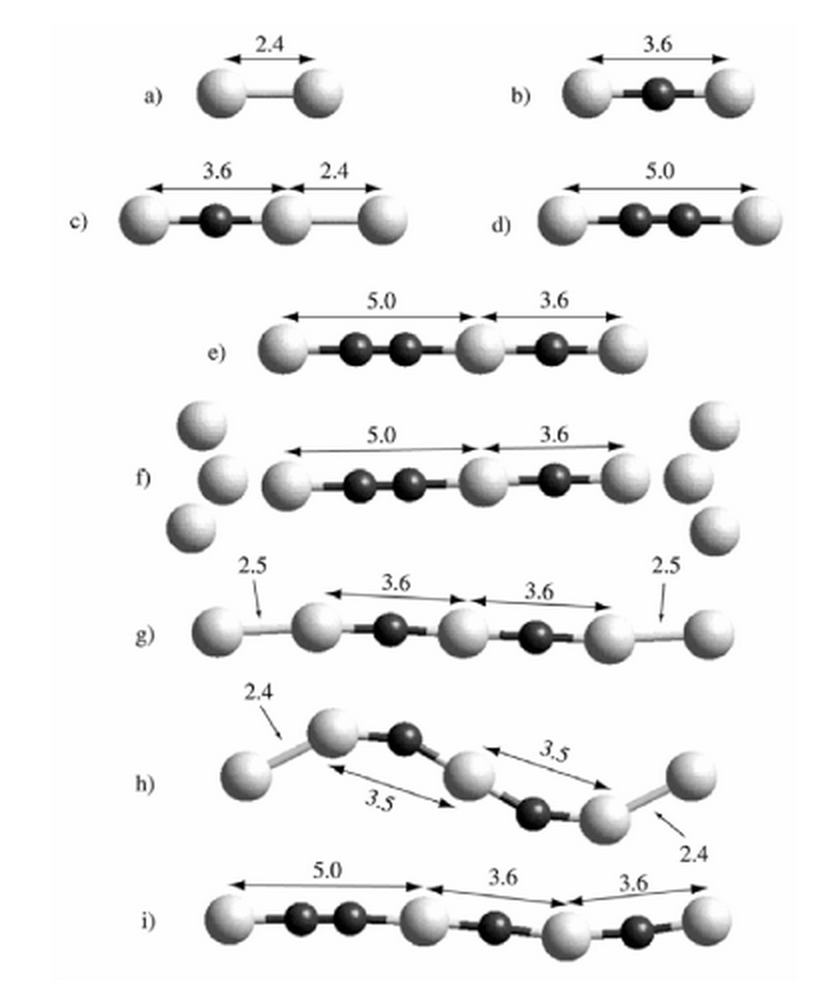
Galvao, Douglas Soares; Rodrigues, Varlei; Ugarte, Daniel; Legoas, Sergio Benites
The role of carbon contamination in metallic nanowires Journal Article
In: Materials Research, vol. 7, no. 2, pp. 339–342, 2004.
Abstract | Links | BibTeX | Tags: Contaminantes, Electronic Structure, Linear Atomic Chains, Metallic Nanowires, Structure
@article{galvao2004role,
title = {The role of carbon contamination in metallic nanowires},
author = {Galvao, Douglas Soares and Rodrigues, Varlei and Ugarte, Daniel and Legoas, Sergio Benites},
url = {http://www.scielo.br/scielo.php?pid=S1516-14392004000200020&script=sci_arttext},
year = {2004},
date = {2004-01-01},
journal = {Materials Research},
volume = {7},
number = {2},
pages = {339--342},
publisher = {SciELO Brasil},
abstract = {Metallic nanowires have attracted much attention in the last years due to new phenomena such as quantum conductance and the existence of unexpected long interatomic distances attaining 0.3-0.5 nm. These large distances represented a challenge for physical interpretation. In this work we present experimental data from high-resolution transmission electron microscopy and results from ab initio calculations for suspended gold chains and show that these large distances can be easily explained by the presence of carbon atoms as contaminants. In principle the present conclusions can be also applied to other metallic nanowires (such as Ag and Pt) whose structures also present large interatomic distances.},
keywords = {Contaminantes, Electronic Structure, Linear Atomic Chains, Metallic Nanowires, Structure},
pubstate = {published},
tppubtype = {article}
}

Coluci, VR; Galvao, DS; Baughman, RH
Theoretical investigation of electromechanical effects for graphyne carbon nanotubes Journal Article
In: The Journal of chemical physics, vol. 121, no. 7, pp. 3228–3237, 2004.
Abstract | Links | BibTeX | Tags: Allotropes, Electroactuation, Electronic Structure, Graphynes, Nanotubes
@article{coluci2004theoretical,
title = {Theoretical investigation of electromechanical effects for graphyne carbon nanotubes},
author = {Coluci, VR and Galvao, DS and Baughman, RH},
url = {http://scitation.aip.org/content/aip/journal/jcp/121/7/10.1063/1.1772756},
year = {2004},
date = {2004-01-01},
journal = {The Journal of chemical physics},
volume = {121},
number = {7},
pages = {3228--3237},
publisher = {AIP Publishing},
abstract = {We present a theoretical study of the electronic and mechanical properties of graphyne-based nanotubes (GNTs). These semiconducting nanotubes result from the elongation of one-third of the covalent interconnections of graphite-based nanotubes by the introduction of yne groups. The effect of charge injection on the dimensions of GNTs was investigated using tight-binding calculations. Low amounts of electron injection are predicted to cause qualitatively different responses for armchair and zigzag graphyne nanotubes. Although the behavior is qualitatively similar to the usual carbon nanotubes, the charge-induced strains are predicted to be smaller for the GNTs than for ordinary single walled carbon nanotubes.},
keywords = {Allotropes, Electroactuation, Electronic Structure, Graphynes, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
2003
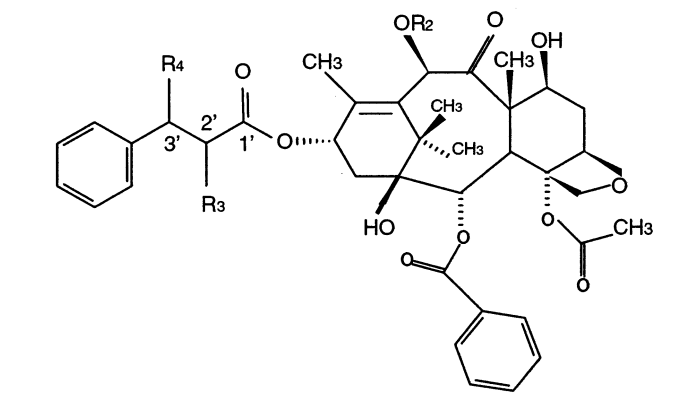
Braga, Scheila Furtado; Galvao, Douglas Soares
A structure-activity study of taxol, taxotere, and derivatives using the electronic indices methodology (EIM) Journal Article
In: Journal of chemical information and computer sciences, vol. 43, no. 2, pp. 699–706, 2003.
Abstract | Links | BibTeX | Tags: Drug Design, Electronic Structure, Taxol, Taxotere, Theory of Electronic Indices
@article{braga2003structure,
title = {A structure-activity study of taxol, taxotere, and derivatives using the electronic indices methodology (EIM)},
author = {Braga, Scheila Furtado and Galvao, Douglas Soares},
url = {http://pubs.acs.org/doi/abs/10.1021/ci025640v},
year = {2003},
date = {2003-01-01},
journal = {Journal of chemical information and computer sciences},
volume = {43},
number = {2},
pages = {699--706},
publisher = {American Chemical Society},
abstract = {Among the new families of effective anticancer drugs, the natural product paclitaxel (Taxol/Bristol-MyersSquibb)
and its semisynthetic derivative docetaxel (Taxotere/Rhone-Poulenc Rorer) are probably the most
promising agents under investigation. Surprisingly considering their importance no detailed quantum
mechanical studies have been carried out for these drugs. In this work we report the first structure-activity
relationship (SAR) studies for 20 taxoid structures using molecular descriptors from all-electron quantum
methods. The used methods were the pattern-recognition Principal Component Analysis (PCA), Hierarchical
Clustering Analysis (HCA), and the recently developed Electronic Indices Methodology (EIM). The combined
use of EIM with PCA/HCA methodologies was able to correctly classify active and inactive taxoids with
100% of accuracy using only a few “universal” quantum molecular descriptors. It was possible to identify
the electronic features defining active molecules. This information can be used to select and design new
active compounds. The combined use of EIM with PCA/HCA can be a new and very efficient tool in the
field of computer assisted drug design.},
keywords = {Drug Design, Electronic Structure, Taxol, Taxotere, Theory of Electronic Indices},
pubstate = {published},
tppubtype = {article}
}
and its semisynthetic derivative docetaxel (Taxotere/Rhone-Poulenc Rorer) are probably the most
promising agents under investigation. Surprisingly considering their importance no detailed quantum
mechanical studies have been carried out for these drugs. In this work we report the first structure-activity
relationship (SAR) studies for 20 taxoid structures using molecular descriptors from all-electron quantum
methods. The used methods were the pattern-recognition Principal Component Analysis (PCA), Hierarchical
Clustering Analysis (HCA), and the recently developed Electronic Indices Methodology (EIM). The combined
use of EIM with PCA/HCA methodologies was able to correctly classify active and inactive taxoids with
100% of accuracy using only a few “universal” quantum molecular descriptors. It was possible to identify
the electronic features defining active molecules. This information can be used to select and design new
active compounds. The combined use of EIM with PCA/HCA can be a new and very efficient tool in the
field of computer assisted drug design.
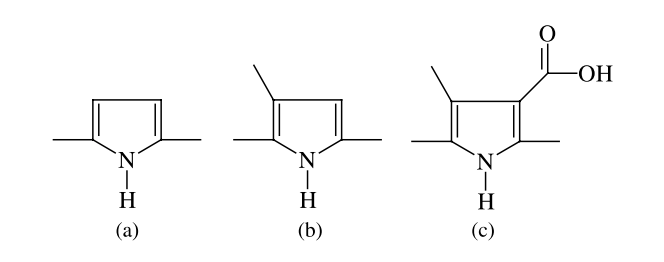
Del Nero, J; Galvao, DS; Laks, B
Electronic structure investigation of biosensor polymer Journal Article
In: Optical Materials, vol. 21, no. 1, pp. 461–466, 2003.
Abstract | Links | BibTeX | Tags: Conducting Polymers, Electronic Structure, Sensors
@article{del2003electronic,
title = {Electronic structure investigation of biosensor polymer},
author = {Del Nero, J and Galvao, DS and Laks, B},
url = {http://www.sciencedirect.com/science/article/pii/S0925346702001830},
year = {2003},
date = {2003-01-01},
journal = {Optical Materials},
volume = {21},
number = {1},
pages = {461--466},
publisher = {Elsevier},
abstract = {We report a theoretical study of the ground, excited and ionic states of 3-methyl pyrrole-4-carboxilic acid (MPC) oligomers and related compounds which present conformational defects. Our results reveal the existence of differentiated electronic behavior for MPC with relation to oligopyrrole derivatives. These electronic features might explain why MPC works properly as a biosensor for cytochrome C while no voltametric response is observed for unsubstituted poly(pyrrole).},
keywords = {Conducting Polymers, Electronic Structure, Sensors},
pubstate = {published},
tppubtype = {article}
}
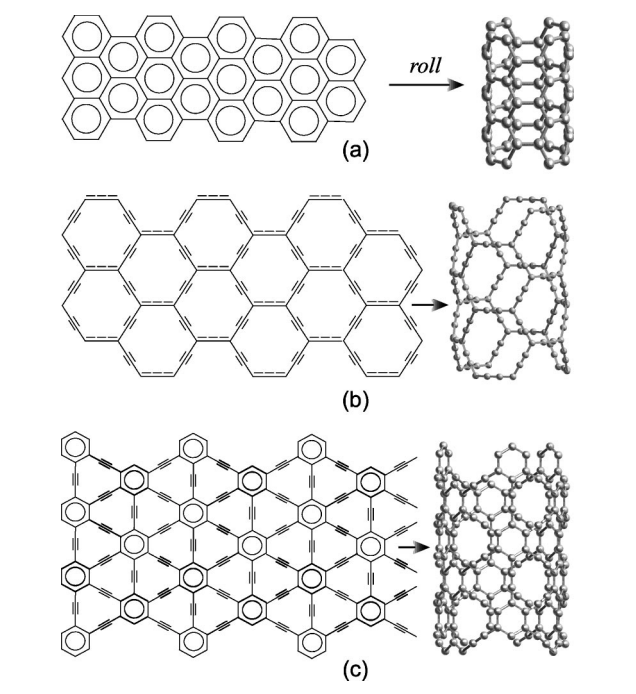
Coluci, VR; Braga, SF; Legoas, SB; Galvao, DS; Baughman, RH
Families of carbon nanotubes: Graphyne-based nanotubes Journal Article
In: Physical Review B, vol. 68, no. 3, pp. 035430, 2003.
Abstract | Links | BibTeX | Tags: Allotropes, Electronic Structure, Graphynes, Nanotubes
@article{coluci2003families,
title = {Families of carbon nanotubes: Graphyne-based nanotubes},
author = {Coluci, VR and Braga, SF and Legoas, SB and Galvao, DS and Baughman, RH},
url = {http://journals.aps.org/prb/abstract/10.1103/PhysRevB.68.035430},
year = {2003},
date = {2003-01-01},
journal = {Physical Review B},
volume = {68},
number = {3},
pages = {035430},
publisher = {APS},
abstract = {New families of carbon single-walled nanotubes are proposed and their electronic structures are investigated. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Analogously to ordinary nanotubes, armchair, zigzag, and chiral graphyne nanotubes are possible. We here predict the electronic properties of these unusual nanotubes using tight-binding and ab initio density functional methods. Of the three graphyne nanotube families analyzed here, two provide metallic behavior for armchair tubes and either metallic or semiconducting behavior for zigzag nanotubes. A diameter- and chirality-independent band gap is predicted for the other investigated graphyne family, as well as an oscillatory dependence of the effective mass on nanotube diameter.},
keywords = {Allotropes, Electronic Structure, Graphynes, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
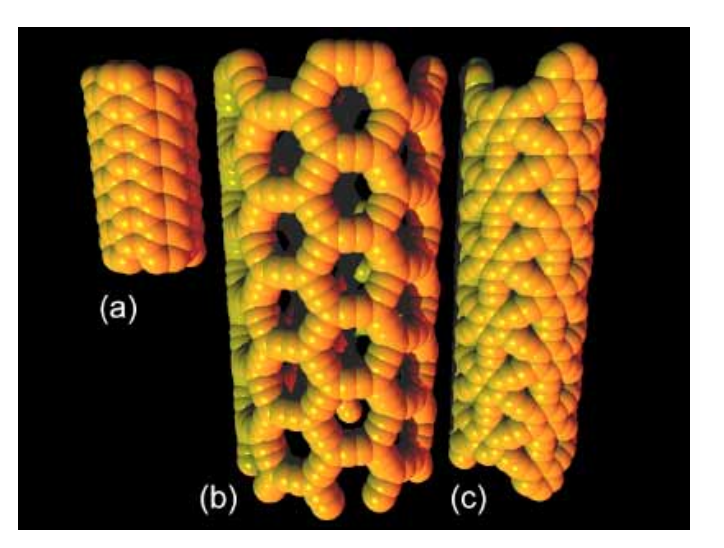
Coluci, VR; Braga, SF; Legoas, SB; Galvao, DS; Baughman, RH
Graphyne Nanotubes: New Families of Carbon Nanotubes Proceedings
Warrendale, Pa.; Materials Research Society; 1999, vol. 739, 2003.
Abstract | Links | BibTeX | Tags: Allotropes, Electronic Structure, Graphynes, Nanotubes
@proceedings{coluci2003graphyne,
title = {Graphyne Nanotubes: New Families of Carbon Nanotubes},
author = {Coluci, VR and Braga, SF and Legoas, SB and Galvao, DS and Baughman, RH},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8031794},
year = {2003},
date = {2003-01-01},
journal = {MATERIALS RESEARCH SOCIETY SYMPOSIUM PROCEEDINGS},
volume = {739},
pages = {175--180},
publisher = {Warrendale, Pa.; Materials Research Society; 1999},
abstract = {Fundamentally new families of carbon single walled nanotubes are proposed. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Similarly to ordinary nanotubes, armchair, zig-zag, and chiral graphyne nanotubes are possible. We present here results for the electronic properties of graphyne based tubes obtained from tight-binding and ab initio density functional methods.},
keywords = {Allotropes, Electronic Structure, Graphynes, Nanotubes},
pubstate = {published},
tppubtype = {proceedings}
}
Giro, R; Galvao, DS
Semiempirical studies of the electronic structure of polyphenylene sulfide phenyleneamine Journal Article
In: International journal of quantum chemistry, vol. 95, no. 3, pp. 252–259, 2003.
Abstract | Links | BibTeX | Tags: co-polymers, Conducting Polymers, Electronic Structure, PANi, PPS, PPSA
@article{giro2003semiempirical,
title = {Semiempirical studies of the electronic structure of polyphenylene sulfide phenyleneamine},
author = {Giro, R and Galvao, DS},
url = {http://onlinelibrary.wiley.com/doi/10.1002/qua.10722/full},
year = {2003},
date = {2003-01-01},
journal = {International journal of quantum chemistry},
volume = {95},
number = {3},
pages = {252--259},
publisher = {Wiley Subscription Services, Inc., A Wiley Company},
abstract = {Polyphenylene sulfide (PPS) and polyaniline (PANI) are heteroatom-containing polymers with unique properties. While PPS provides a good level of chemical and thermal stability, PANI is important due to its ability to form electrically conducting films. A combination of PPS and PANI, with alternating phenyleneamine and phenylene sulfide blocks, resulted in a new material combining the structural features of PPS and PANI. This copolymer is known as polyphenylene sulfide–phenyleneamine (PPSA). In this work we present geometric and spectroscopic studies on PPSA oligomers using the well-known semiempirical methods PM3 (parametric method 3) and ZINDO-S/CI (Zerner's intermediate neglect of differential overlap/spectroscopic-configuration interaction). PM3 results show that long PPSA oligomers present alternating in- and out-of-plane rings with torsion angles about 60°. The ZINDO-simulated spectra compare well with the available experimental data. The origin of the low PPSA conductivity is addressed in terms of electronic features presented by isolated polymeric chains. © 2003 Wiley Periodicals, Inc. Int J Quantum Chem 95: 252–259, 2003},
keywords = {co-polymers, Conducting Polymers, Electronic Structure, PANi, PPS, PPSA},
pubstate = {published},
tppubtype = {article}
}
2002
Braga, SF; Galvao, DS
A semiempirical study on the electronic structure of 10-deacetylbaccatin-III Journal Article
In: Journal of Molecular Graphics and Modelling, vol. 21, no. 1, pp. 57–70, 2002.
Abstract | Links | BibTeX | Tags: Baccatin, Drug Design, Electronic Structure, Taxol, Taxotere, Theory of Electronic Indices
@article{braga2002semiempirical,
title = {A semiempirical study on the electronic structure of 10-deacetylbaccatin-III},
author = {Braga, SF and Galvao, DS},
url = {http://www.sciencedirect.com/science/article/pii/S1093326302001213},
year = {2002},
date = {2002-01-01},
journal = {Journal of Molecular Graphics and Modelling},
volume = {21},
number = {1},
pages = {57--70},
publisher = {Elsevier},
abstract = {We performed a conformational and electronic analysis for 10-deacetylbaccatin-III (DBAC) using well-known semiempirical methods (parametric method 3 (PM3) and Zerner’s intermediate neglect of differential overlap (ZINDO)) coupled to the concepts of total and local density of states (LDOS). Our results indicate that regions presented by paclitaxel (Taxol®) as important for the biological activity can be traced out by the electronic features present in DBAC. These molecules differ only by a phenylisoserine side chain. Compared to paclitaxel, DBAC has a simpler structure in terms of molecular size and number of degrees of freedom (d.f.). This makes DBAC a good candidate for a preliminary investigation of the taxoid family. Our results question the importance of the oxetane group, which seems to be consistent with recent experimental data.
},
keywords = {Baccatin, Drug Design, Electronic Structure, Taxol, Taxotere, Theory of Electronic Indices},
pubstate = {published},
tppubtype = {article}
}
Giro, R; Cyrillo, M; Galvao, DS
Designing conducting polymers using genetic algorithms Journal Article
In: Chemical Physics Letters, vol. 366, no. 1, pp. 170–175, 2002.
Abstract | Links | BibTeX | Tags: Conducting Polymers, Electronic Structure, Genetic Algorithms, Material Design
@article{giro2002designing,
title = {Designing conducting polymers using genetic algorithms},
author = {Giro, R and Cyrillo, M and Galvao, DS},
url = {http://www.sciencedirect.com/science/article/pii/S0009261402015476},
year = {2002},
date = {2002-01-01},
journal = {Chemical Physics Letters},
volume = {366},
number = {1},
pages = {170--175},
publisher = {North-Holland},
abstract = {We have developed a new methodology to design conducting polymers with pre-specified properties. The methodology is based on the use of genetic algorithms (GAs) coupled to Negative Factor Counting technique. We present the results for a case study of polyanilines, one of the most important families of conducting polymers. The methodology proved to be able of generating automatic solutions for the problem of determining the optimum relative concentration for binary and ternary disordered polyaniline alloys exhibiting metallic properties. The methodology is completely general and can be used to design new classes of materials.},
keywords = {Conducting Polymers, Electronic Structure, Genetic Algorithms, Material Design},
pubstate = {published},
tppubtype = {article}
}
Galvao, DS; Braga, SF; Barone, PMVB; Dantas, SO
Dual-Mode Optical Molecular Switching Systems for Organic Memories Proceedings
Cambridge Univ Press, vol. 708, 2002.
Abstract | Links | BibTeX | Tags: Electronic Structure, Molecular Electronics
@proceedings{galvao2002dual,
title = {Dual-Mode Optical Molecular Switching Systems for Organic Memories},
author = {Galvao, DS and Braga, SF and Barone, PMVB and Dantas, SO},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8214320},
year = {2002},
date = {2002-01-01},
journal = {MATERIALS RESEARCH SOCIETY SYMPOSIUM PROCEEDINGS},
volume = {708},
pages = {335--340},
publisher = {Cambridge Univ Press},
abstract = {The synthesis of dual-mode optical molecular switching systems has been recently achieved. These systems were based on chiral helical-shaped alkenes in which the chirality can be reversibly modulated by light. In this work we report a theoretical study on the geometric and spectroscopic properties of these structures using the well-known semi-empirical methods PM3 (Parametric Method 3) and ZINDO/S-CI (Zerner's Intermediate Neglect of Differential Overlap -Spectroscopic - Configuration Interaction). Our results show that there are two stable conformers very close in energy for each possible molecular helicity presenting a barrier of ∼40 kcal/mol for bond rotation along the main molecular axis. Under protonation these barriers increase significantly and might explain why the protonation leads to the blocking of the switching process. We propose a scheme for the switching mechanism based on charge transfer and conformational changes during the isomer interconversion.},
keywords = {Electronic Structure, Molecular Electronics},
pubstate = {published},
tppubtype = {proceedings}
}
Coluci, V R; Braga, SF; Legoas, Sergio B; Galvao, Douglas S; Baughman, RH
New families of carbon nanotubes Journal Article
In: arXiv preprint cond-mat/0207085, 2002.
Abstract | Links | BibTeX | Tags: Allotropes, Electronic Structure, Graphynes, Nanotubes
@article{coluci2002new,
title = {New families of carbon nanotubes},
author = {Coluci, V R and Braga, SF and Legoas, Sergio B and Galvao, Douglas S and Baughman, RH},
url = {http://arxiv.org/abs/cond-mat/0207085},
year = {2002},
date = {2002-01-01},
journal = {arXiv preprint cond-mat/0207085},
abstract = {Fundamentally new families of carbon single walled nanotubes are proposed. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Similarly to ordinary nanotubes, arm-chair, zig-zag, and chiral graphyne nanotubes are possible. Electronic properties, predicted using tight-binding and ab initio density functional methods, show a rich variety of metallic and semiconducting behaviors.},
keywords = {Allotropes, Electronic Structure, Graphynes, Nanotubes},
pubstate = {published},
tppubtype = {article}
}

Legoas, Sergio B; Galvao, Douglas S; Rodrigues, Varlei; Ugarte, Daniel
Origin of anomalously long interatomic distances in suspended gold chains Journal Article
In: Physical Review Letters, vol. 88, no. 7, pp. 076105, 2002.
Abstract | Links | BibTeX | Tags: DFT, Electronic Structure, Linear Atomic Chains, Metallic Nanowires, TEM, top20
@article{legoas2002origin,
title = {Origin of anomalously long interatomic distances in suspended gold chains},
author = {Legoas, Sergio B and Galvao, Douglas S and Rodrigues, Varlei and Ugarte, Daniel},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.88.076105},
year = {2002},
date = {2002-01-01},
journal = {Physical Review Letters},
volume = {88},
number = {7},
pages = {076105},
publisher = {American Physical Society},
abstract = {The discovery of long bonds in gold atom chains has represented a challenge for physical interpretation. In fact, interatomic distances frequently attain 3.0–3.6 Å values, and distances as large as 5.0 Å may be occasionally observed. Here we studied gold chains by transmission electron microscopy and performed theoretical calculations using cluster ab initio density functional formalism. We show that the insertion of two carbon atoms is required to account for the longest bonds, while distances above 3 Å may be due to a mixture of clean and one C atom contaminated bonds.},
keywords = {DFT, Electronic Structure, Linear Atomic Chains, Metallic Nanowires, TEM, top20},
pubstate = {published},
tppubtype = {article}
}
Vendrame, R; Coluci, VR; Braga, RS; Galvao, DS
Structure--activity relationship (SAR) studies of the tripos benchmark steroids Journal Article
In: Journal of Molecular Structure: THEOCHEM, vol. 619, no. 1, pp. 195–205, 2002.
Abstract | Links | BibTeX | Tags: Drug Design, Electronic Structure, HCA/PCA, Neural Networks, Tripos
@article{vendrame2002structure,
title = {Structure--activity relationship (SAR) studies of the tripos benchmark steroids},
author = {Vendrame, R and Coluci, VR and Braga, RS and Galvao, DS},
url = {http://www.sciencedirect.com/science/article/pii/S016612800200578X},
year = {2002},
date = {2002-01-01},
journal = {Journal of Molecular Structure: THEOCHEM},
volume = {619},
number = {1},
pages = {195--205},
publisher = {Elsevier},
abstract = {We report here qualitative structure–activity relationship (SAR) studies for the molecular set called Tripos or Cramer steroid data set. These compounds are known to bind to corticosteroid binding globulin (CBG). In the present work we have used the electronic indices methodology (EIM). The EIM is based on Boolean relational rules exploring the concepts of local density of states and critical values for energy separation involving frontier orbitals. We have also carried out comparative principal component analysis (PCA) and hierarchical clustering analysis (HCA) studies with molecular descriptors obtained from EIM calculations. EIM, PCA and HCA correctly predict (100% accuracy) the steroid's biological activity. The present studies reinforce the universal applicability of the EIM descriptors and show that the combined use of EIM coupled to PCA can be a new efficient and powerful SAR tool.
},
keywords = {Drug Design, Electronic Structure, HCA/PCA, Neural Networks, Tripos},
pubstate = {published},
tppubtype = {article}
}
Legoas, Sergio B; Galvao, Douglas S; Rodrigues, Varlei; Ugarte, Daniel
The Role of Carbon Contamination in Suspended Gold Nanowires Journal Article
In: MRS Proceedings, vol. 738, pp. G14–6, 2002.
Abstract | Links | BibTeX | Tags: DFT, Electronic Structure, Linear Atomic Chains, Metallic Nanowires, TEM
@article{legoas2002role,
title = {The Role of Carbon Contamination in Suspended Gold Nanowires},
author = {Legoas, Sergio B and Galvao, Douglas S and Rodrigues, Varlei and Ugarte, Daniel},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8031754&fileId=S1946427400148249},
year = {2002},
date = {2002-01-01},
journal = {MRS Proceedings},
volume = {738},
pages = {G14--6},
publisher = {Cambridge University Press},
abstract = {Metallic nanowires represent very interesting systems due to new phenomena such as quantum conductance and unexpected long interatomic distances attaining 0.3–0.5 nm. These large distances represent a challenge for physical interpretation. In this work we present experimental data from transmission electron microscopy and results from ab initio density functional calculations for suspended gold chains. We show that large distances as 0.5 nm can be easily explained by the presence of carbon atoms as contaminants, while distances ranging from 0.29 up to 0.36 nm might be explained as resulting of a mixture of clean stressed and contaminated linear chains.},
keywords = {DFT, Electronic Structure, Linear Atomic Chains, Metallic Nanowires, TEM},
pubstate = {published},
tppubtype = {article}
}
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ

