
Nadia F. Andradea Gustavo Brunettoa, Douglas S. Galvao
High Pressure Induced Binding Between Linear Carbon Chains and Nanotubes Proceedings
vol. 1752, no. 53-58, 2015, (MRS Proceedings, 1752, pp 53-58).
@proceedings{Brunettoa2015,
title = {High Pressure Induced Binding Between Linear Carbon Chains and Nanotubes},
author = {Gustavo Brunettoa, Nadia F. Andradea, Douglas S. Galvao, Antonio G. Souza Filho},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=9553206&fileId=S1946427415000913},
doi = {10.1557/opl.2015.91},
year = {2015},
date = {2015-01-01},
volume = {1752},
number = {53-58},
abstract = {Recent studies of single-walled carbon nanotubes (CNTs) in aqueous media have showed that water can significantly affect the tube mechanical properties. CNTs under hydrostatic compression can preserve their elastic properties up to large pressure values, while exhibiting exceptional resistance to mechanical loadings. It was experimentally observed that CNTs with encapsulated linear carbon chains (LCCs), when subjected to high hydrostatic pressure values, present irreversible red shifts in some of their vibrational frequencies. In order to address the cause of this phenomenon, we have carried out fully atomistic reactive (ReaxFF) molecular dynamics (MD) simulations for model structures mimicking the experimental conditions. We have considered the cases of finite and infinite (cyclic boundary conditions) CNTs filled with LCCs (LCC@CNTs) of different lengths (from 9 up to 40 atoms). Our results show that increasing the hydrostatic pressure causes the CNT to be deformed in an inhomogeneous way due to the LCC presence. The LCC/CNT interface regions exhibit convex curvatures, which results in more reactive sites, thus favoring the formation of covalent chemical bonds between the chain and the nanotube. This process is irreversible with the newly formed bonds continuing to exist even after releasing the external pressure and causing an irreversibly red shift in the chain vibrational modes from 1850 to 1500 cm−1.},
note = {MRS Proceedings, 1752, pp 53-58},
keywords = {},
pubstate = {published},
tppubtype = {proceedings}
}
Legoas, SB; Dos Santos, RPB; Troche, KS; Coluci, VR; Galvao, DS
Ordered phases of encapsulated diamondoids into carbon nanotubes Journal Article
In: Nanotechnology, vol. 22, no. 31, pp. 315708, 2011.
@article{legoas2011ordered,
title = {Ordered phases of encapsulated diamondoids into carbon nanotubes},
author = {Legoas, SB and Dos Santos, RPB and Troche, KS and Coluci, VR and Galvao, DS},
url = {http://iopscience.iop.org/0957-4484/22/31/315708},
year = {2011},
date = {2011-01-01},
journal = {Nanotechnology},
volume = {22},
number = {31},
pages = {315708},
publisher = {IOP Publishing},
abstract = {Diamondoids are hydrogen-terminated nanosized diamond fragments that are present in petroleum crude oil at low concentrations. These fragments are found as oligomers of the smallest diamondoid, adamantane (C10H16). Due to their small size, diamondoids can be encapsulated into carbon nanotubes to form linear arrangements. We have investigated the encapsulation of diamondoids into single walled carbon nanotubes with diameters between 1.0 and 2.2 nm using fully atomistic simulations. We performed classical molecular dynamics and energy minimizations calculations to determine the most stable configurations. We observed molecular ordered phases (e.g. double, triple, 4- and 5-stranded helices) for the encapsulation of adamantane, diamantane, and dihydroxy diamantane. Our results also indicate that the functionalization of diamantane with hydroxyl groups can lead to an improvement on the molecular packing factor when compared to non-functionalized compounds. Comparisons to hard-sphere models revealed differences, especially when more asymmetrical diamondoids were considered. For larger diamondoids (i.e., adamantane tetramers), we have not observed long-range ordering but only a tendency to form incomplete helical structures. Our calculations predict that thermally stable (at least up to room temperature) complex ordered phases of diamondoids can be formed through encapsulation into carbon nanotubes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Azevedo, David L; Sato, Fernando; Gomes de Sousa Filho, Antonio; Galvao, Douglas S
In: Molecular Simulation, vol. 37, no. 9, pp. 746–751, 2011.
@article{azevedo2011van,
title = {van der Waals potential barrier for cobaltocene encapsulation into single-walled carbon nanotubes: classical molecular dynamics and ab initio study},
author = {Azevedo, David L and Sato, Fernando and Gomes de Sousa Filho, Antonio and Galvao, Douglas S},
url = {http://www.tandfonline.com/doi/abs/10.1080/08927022.2010.537093#.VLfBForF-2o},
year = {2011},
date = {2011-01-01},
journal = {Molecular Simulation},
volume = {37},
number = {9},
pages = {746--751},
publisher = {Taylor & Francis Group},
abstract = {In this work, we carried out geometry optimisations and classical molecular dynamics for the problem of cobaltocene (CC) encapsulation into different carbon nanotubes (CNTs) ((7,7), (8,8), (13,0) and (14,0) tubes were used). CCs are molecules composed of two aromatic pentagonal rings (C5H5) sandwiching one cobalt atom. From our simulation results, we observed that CC was encapsulated into CNTs (8,8), (13,0) and (14,0). However, for CNT (7,7), the encapsulation could not occur, in disaggrement with some previous works in the literature. Our results show that the encapsulation process is mainly governed by van der Waals potential barriers.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Moreira, E; Lemos, V; Galvao, DS; Azevedo, DL
$beta$-Carotene encapsulation into single-walled carbon nanotubes: a theoretical study Journal Article
In: Molecular Simulation, vol. 36, no. 13, pp. 1031–1034, 2010.
@article{moreira2010beta,
title = {$beta$-Carotene encapsulation into single-walled carbon nanotubes: a theoretical study},
author = {Moreira, E and Lemos, V and Galvao, DS and Azevedo, DL},
url = {http://www.tandfonline.com/doi/abs/10.1080/08927022.2010.501519#.VLfmM4rF-2o},
year = {2010},
date = {2010-01-01},
journal = {Molecular Simulation},
volume = {36},
number = {13},
pages = {1031--1034},
publisher = {Taylor & Francis},
abstract = {Recently, the encapsulation of β-carotene molecules into carbon nanotubes has been achieved. In this work, we report molecular dynamics simulations and tight-binding density functional-based results for a theoretical study of the encapsulation processes. Our results show that the molecules undergo geometrical deformations when encapsulated with significant changes in their electronic structure. Based on these results, we propose a new interpretation to the changes associated with the β-carotene absorption bands experimentally observed.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Troche, Karla S; Coluci, Vitor R; Galvao, Douglas S
Atomistic study of the encapsulation of diamondoids inside carbon nanotubes Journal Article
In: arXiv preprint arXiv:0707.1777, 2007.
@article{troche2007atomistic,
title = {Atomistic study of the encapsulation of diamondoids inside carbon nanotubes},
author = {Troche, Karla S and Coluci, Vitor R and Galvao, Douglas S},
url = {http://arxiv.org/abs/0707.1777},
year = {2007},
date = {2007-01-01},
journal = {arXiv preprint arXiv:0707.1777},
abstract = {The encapsulation of hydrogen-terminated nanosized diamond fragments (the so-called diamondoids) into armchair single walled carbon nanotubes with diameters in the range of 1.0 up to 2.2 nm has been investigated using classical molecular dynamics simulations. Diameter dependent molecular ordered phases were found for the encapsulation of adamantane (C10H16), diamantane (C14H20), and dihydroxy diamantane (C14H20O2). The same types of chiral ordered phases (double, triple, 4- and 5-stranded helices) observed for the encapsulation of C60 molecules were also observed for diamondoids. On the other hand, some achiral phases comprising layered structures were not observed. Our results also indicate that the modification of diamantane through functionalization with hydroxyl groups can lead to an enhancement of the packing of molecules inside the nanotubes compared to nonfunctionalized compounds. Comparisons to hard-sphere models are also presented revealing differences, specially when more asymmetrical diamondoids are considered. For larger structures (adamantane tetramers) we have not observed long-range ordering for nanotubes with diameters in the range of 1.49 to 2.17 nm but only a tendency to form incomplete helical structures.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Azevedo, David L; Sato, Fernando; Galvao, Douglas S; others,
Cobaltocene encapsulation into single-walled carbon nanotubes: A molecular dynamics investigation Journal Article
In: arXiv preprint arXiv:0707.3831, 2007.
@article{azevedo2007cobaltocene,
title = {Cobaltocene encapsulation into single-walled carbon nanotubes: A molecular dynamics investigation},
author = {Azevedo, David L and Sato, Fernando and Galvao, Douglas S and others},
url = {http://arxiv.org/abs/0707.3831},
year = {2007},
date = {2007-01-01},
journal = {arXiv preprint arXiv:0707.3831},
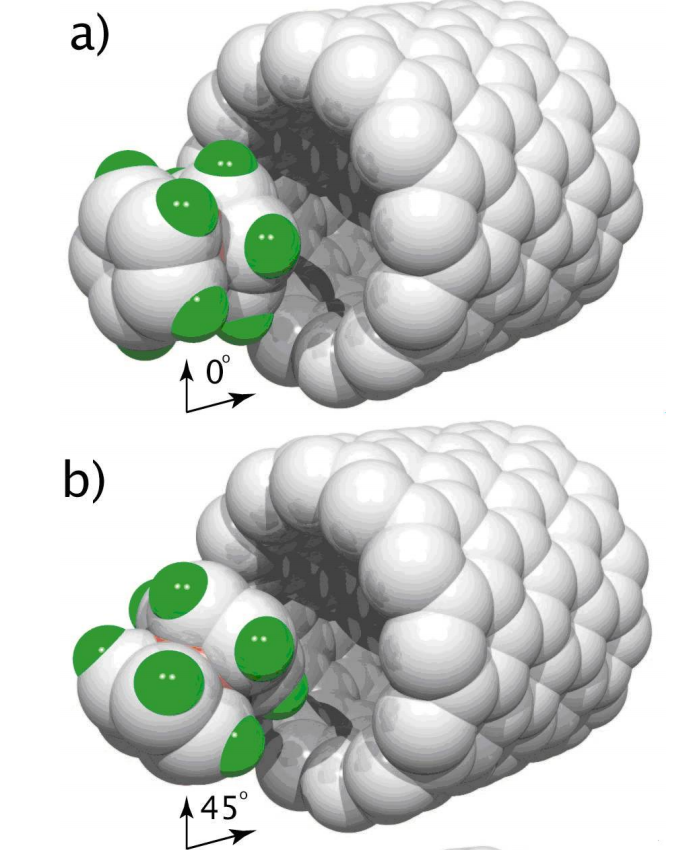
abstract = {Recently (PRL 96, 106804 (2006)) it was suggested that cobaltocene(CC) molecules encapsulated into (7,7) carbon nanotubes (CNT@(7,7)) could be the basis for new spintronic devices. We show here based on impact molecular dynamics and DFT calculations that when dynamical aspects are explicitly considered the CC encapsulation into CNT@(7,7) does not occur, it is prevented by a dynamic barrier mainly due to van der Waals interactions. Our results show that CNT@(13,0) having enough axial space for encapsulation but no enough one to allow freely rotation of the cobaltocene molecule would be a feasible candidate to such application.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Troche, KS; Coluci, VR; Rurali, R; Galvao, DS
Structural and electronic properties of zigzag carbon nanotubes filled with small fullerenes Journal Article
In: Journal of Physics: Condensed Matter, vol. 19, no. 23, pp. 236222, 2007.
@article{troche2007structural,
title = {Structural and electronic properties of zigzag carbon nanotubes filled with small fullerenes},
author = {Troche, KS and Coluci, VR and Rurali, R and Galvao, DS},
url = {http://iopscience.iop.org/0953-8984/19/23/236222},
year = {2007},
date = {2007-01-01},
journal = {Journal of Physics: Condensed Matter},
volume = {19},
number = {23},
pages = {236222},
publisher = {IOP Publishing},
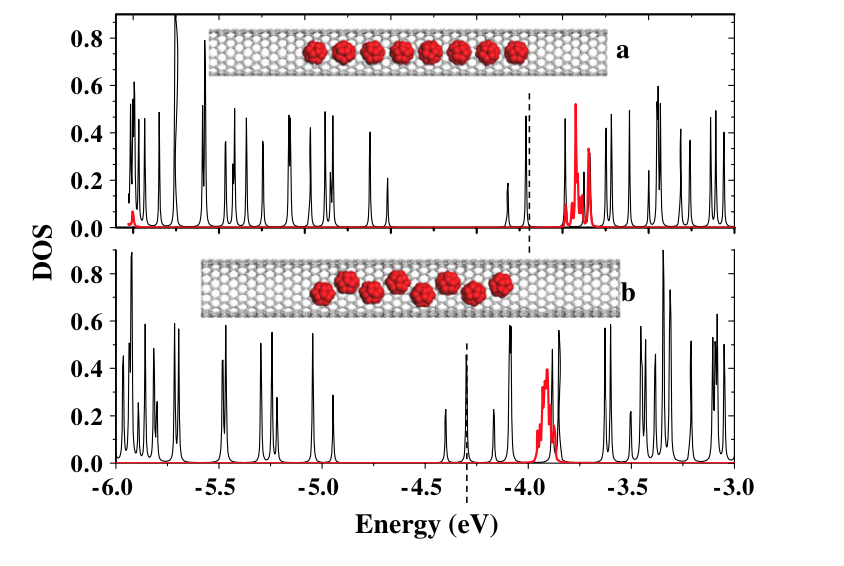
abstract = {In this work we investigated the encapsulation of C20 and C30 fullerenes into semiconducting carbon nanotubes to study the possibility of bandgap engineering in such systems. Classical molecular dynamics simulations coupled to tight-binding calculations were used to determine the conformational and electronic properties of carbon nanotubes with an increasing fullerene concentration. We have observed that C20 fullerenes behave similarly to a n-type dopant while C30 can provide p-type doping in some cases. The combined incorporation of both types of fullerenes (hybrid encapsulation) into the same nanotube leads to a behaviour similar to that found in electronic pn-junctions. These aspects can be exploited in the design of nanoelectronic devices using semiconducting carbon nanotubes.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Troche, KS; Coluci, VR; Rurali, R; Galvao, DS
Doping of zigzag carbon nanotubes through the encapsulation of small fullerenes Journal Article
In: arXiv preprint cond-mat/0607197, 2006.
@article{troche2006doping,
title = {Doping of zigzag carbon nanotubes through the encapsulation of small fullerenes},
author = {Troche, KS and Coluci, VR and Rurali, R and Galvao, DS},
url = {http://arxiv.org/abs/cond-mat/0607197},
year = {2006},
date = {2006-01-01},
journal = {arXiv preprint cond-mat/0607197},
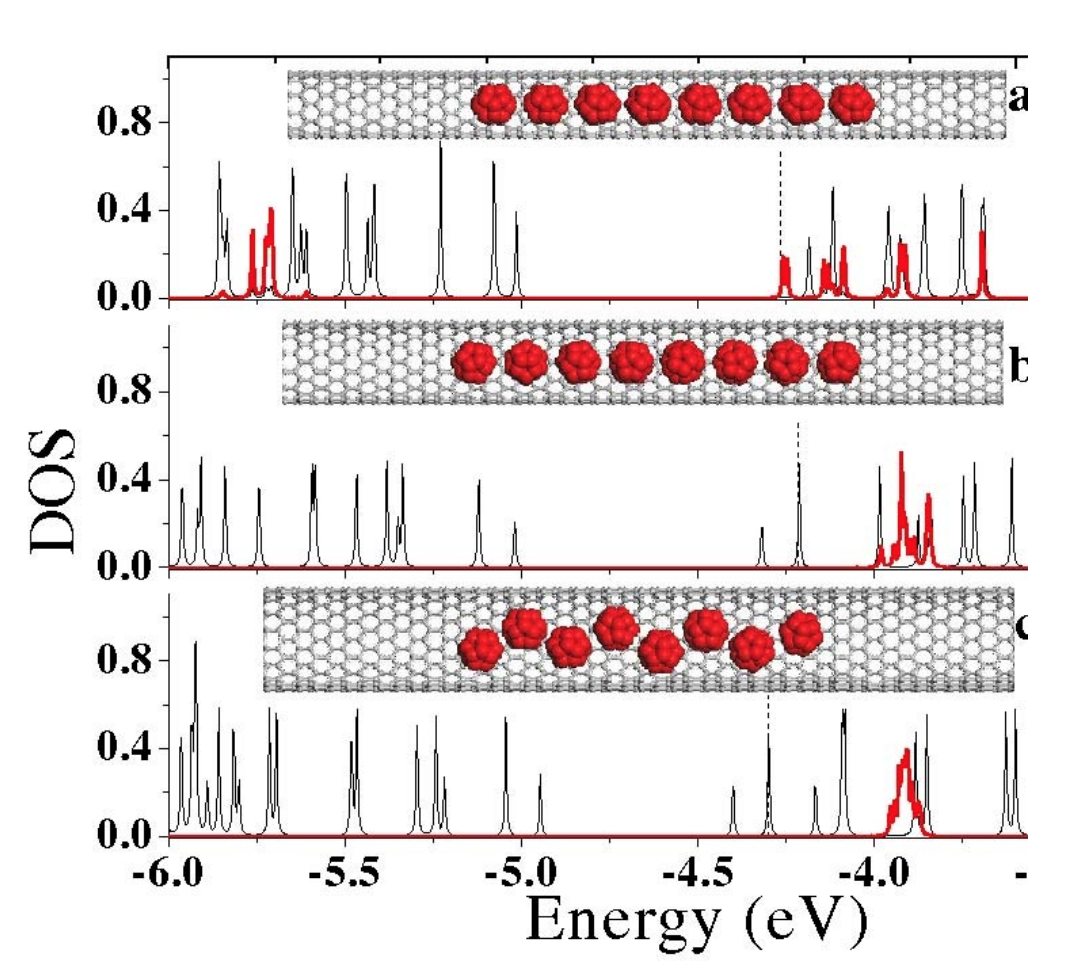
abstract = {In this work we investigated the encapsulation of C20 and C30 fullerenes into semiconducting carbon nanotubes to study the possibility of bandgap engineering in such systems. Classical molecular dynamics simulations coupled to tight-binding calculations were used to determine the conformational and electronic properties of carbon nanotube supercells containing up to 12 fullerenes. We have observed that C20 fullerenes behave similarly to a p-type dopant while C30 ones work as n-type ones. For larger diameter nanotubes, where fullerene patterns start to differ from the linear arrangements (peapods), the doping features are preserved for both fullerenes, but local disorder plays an important role and significantly alters the electronic structure. The combined incorporation of both fullerene types (hybrid encapsulation) into the same nanotube leads to a behavior similar to that found in electronic junctions in Silicon-based electronic devices. These aspects can be exploited in the design of nanoelectronic devices using semiconducting carbon nanotubes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2015

Nadia F. Andradea Gustavo Brunettoa, Douglas S. Galvao
High Pressure Induced Binding Between Linear Carbon Chains and Nanotubes Proceedings
vol. 1752, no. 53-58, 2015, (MRS Proceedings, 1752, pp 53-58).
Abstract | Links | BibTeX | Tags: CNT encapsulation, Electronic Structure, Linear Chains, Molecular Dynamics
@proceedings{Brunettoa2015,
title = {High Pressure Induced Binding Between Linear Carbon Chains and Nanotubes},
author = {Gustavo Brunettoa, Nadia F. Andradea, Douglas S. Galvao, Antonio G. Souza Filho},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=9553206&fileId=S1946427415000913},
doi = {10.1557/opl.2015.91},
year = {2015},
date = {2015-01-01},
volume = {1752},
number = {53-58},
abstract = {Recent studies of single-walled carbon nanotubes (CNTs) in aqueous media have showed that water can significantly affect the tube mechanical properties. CNTs under hydrostatic compression can preserve their elastic properties up to large pressure values, while exhibiting exceptional resistance to mechanical loadings. It was experimentally observed that CNTs with encapsulated linear carbon chains (LCCs), when subjected to high hydrostatic pressure values, present irreversible red shifts in some of their vibrational frequencies. In order to address the cause of this phenomenon, we have carried out fully atomistic reactive (ReaxFF) molecular dynamics (MD) simulations for model structures mimicking the experimental conditions. We have considered the cases of finite and infinite (cyclic boundary conditions) CNTs filled with LCCs (LCC@CNTs) of different lengths (from 9 up to 40 atoms). Our results show that increasing the hydrostatic pressure causes the CNT to be deformed in an inhomogeneous way due to the LCC presence. The LCC/CNT interface regions exhibit convex curvatures, which results in more reactive sites, thus favoring the formation of covalent chemical bonds between the chain and the nanotube. This process is irreversible with the newly formed bonds continuing to exist even after releasing the external pressure and causing an irreversibly red shift in the chain vibrational modes from 1850 to 1500 cm−1.},
note = {MRS Proceedings, 1752, pp 53-58},
keywords = {CNT encapsulation, Electronic Structure, Linear Chains, Molecular Dynamics},
pubstate = {published},
tppubtype = {proceedings}
}
2011
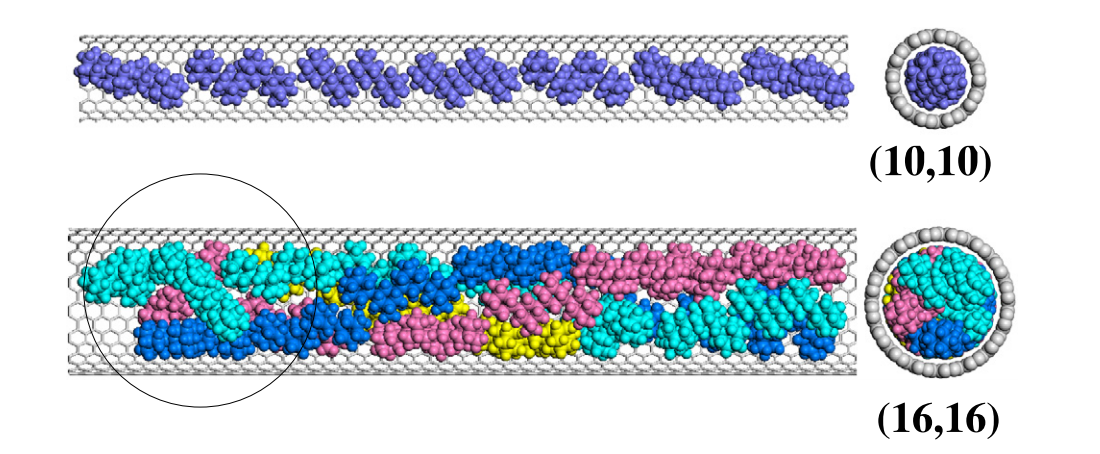
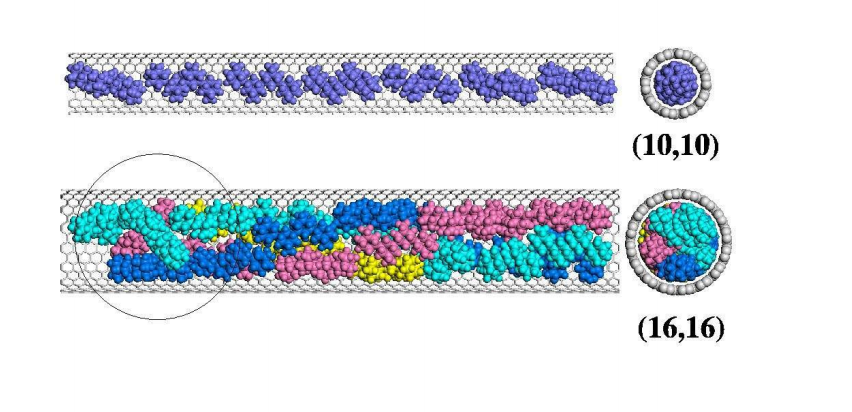
Legoas, SB; Dos Santos, RPB; Troche, KS; Coluci, VR; Galvao, DS
Ordered phases of encapsulated diamondoids into carbon nanotubes Journal Article
In: Nanotechnology, vol. 22, no. 31, pp. 315708, 2011.
Abstract | Links | BibTeX | Tags: CNT encapsulation, Diamondoids, Molecular Dynamics
@article{legoas2011ordered,
title = {Ordered phases of encapsulated diamondoids into carbon nanotubes},
author = {Legoas, SB and Dos Santos, RPB and Troche, KS and Coluci, VR and Galvao, DS},
url = {http://iopscience.iop.org/0957-4484/22/31/315708},
year = {2011},
date = {2011-01-01},
journal = {Nanotechnology},
volume = {22},
number = {31},
pages = {315708},
publisher = {IOP Publishing},
abstract = {Diamondoids are hydrogen-terminated nanosized diamond fragments that are present in petroleum crude oil at low concentrations. These fragments are found as oligomers of the smallest diamondoid, adamantane (C10H16). Due to their small size, diamondoids can be encapsulated into carbon nanotubes to form linear arrangements. We have investigated the encapsulation of diamondoids into single walled carbon nanotubes with diameters between 1.0 and 2.2 nm using fully atomistic simulations. We performed classical molecular dynamics and energy minimizations calculations to determine the most stable configurations. We observed molecular ordered phases (e.g. double, triple, 4- and 5-stranded helices) for the encapsulation of adamantane, diamantane, and dihydroxy diamantane. Our results also indicate that the functionalization of diamantane with hydroxyl groups can lead to an improvement on the molecular packing factor when compared to non-functionalized compounds. Comparisons to hard-sphere models revealed differences, especially when more asymmetrical diamondoids were considered. For larger diamondoids (i.e., adamantane tetramers), we have not observed long-range ordering but only a tendency to form incomplete helical structures. Our calculations predict that thermally stable (at least up to room temperature) complex ordered phases of diamondoids can be formed through encapsulation into carbon nanotubes.},
keywords = {CNT encapsulation, Diamondoids, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
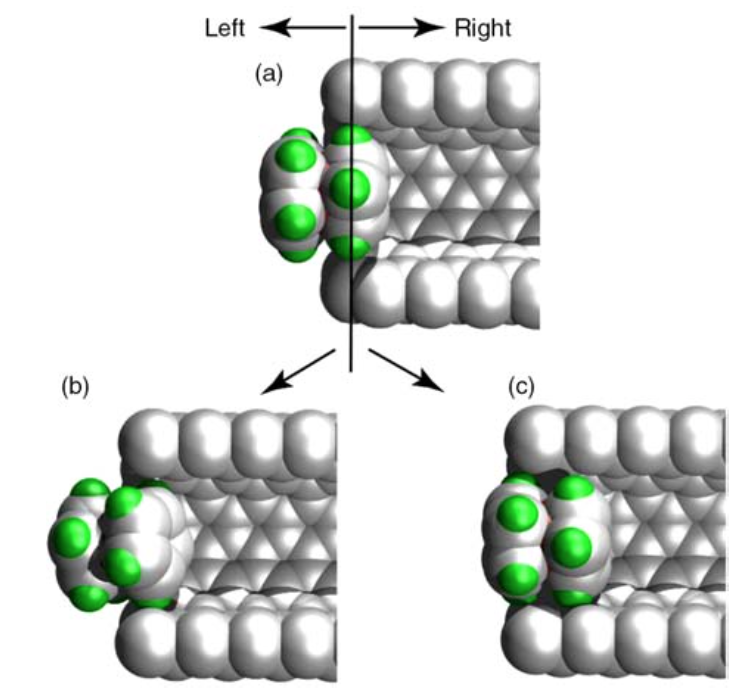
Azevedo, David L; Sato, Fernando; Gomes de Sousa Filho, Antonio; Galvao, Douglas S
In: Molecular Simulation, vol. 37, no. 9, pp. 746–751, 2011.
Abstract | Links | BibTeX | Tags: CNT encapsulation, Cobaltocene, Molecular Dynamics
@article{azevedo2011van,
title = {van der Waals potential barrier for cobaltocene encapsulation into single-walled carbon nanotubes: classical molecular dynamics and ab initio study},
author = {Azevedo, David L and Sato, Fernando and Gomes de Sousa Filho, Antonio and Galvao, Douglas S},
url = {http://www.tandfonline.com/doi/abs/10.1080/08927022.2010.537093#.VLfBForF-2o},
year = {2011},
date = {2011-01-01},
journal = {Molecular Simulation},
volume = {37},
number = {9},
pages = {746--751},
publisher = {Taylor & Francis Group},
abstract = {In this work, we carried out geometry optimisations and classical molecular dynamics for the problem of cobaltocene (CC) encapsulation into different carbon nanotubes (CNTs) ((7,7), (8,8), (13,0) and (14,0) tubes were used). CCs are molecules composed of two aromatic pentagonal rings (C5H5) sandwiching one cobalt atom. From our simulation results, we observed that CC was encapsulated into CNTs (8,8), (13,0) and (14,0). However, for CNT (7,7), the encapsulation could not occur, in disaggrement with some previous works in the literature. Our results show that the encapsulation process is mainly governed by van der Waals potential barriers.},
keywords = {CNT encapsulation, Cobaltocene, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
2010
Moreira, E; Lemos, V; Galvao, DS; Azevedo, DL
$beta$-Carotene encapsulation into single-walled carbon nanotubes: a theoretical study Journal Article
In: Molecular Simulation, vol. 36, no. 13, pp. 1031–1034, 2010.
Abstract | Links | BibTeX | Tags: Beta-carotene, CNT encapsulation, Molecular Dynamics
@article{moreira2010beta,
title = {$beta$-Carotene encapsulation into single-walled carbon nanotubes: a theoretical study},
author = {Moreira, E and Lemos, V and Galvao, DS and Azevedo, DL},
url = {http://www.tandfonline.com/doi/abs/10.1080/08927022.2010.501519#.VLfmM4rF-2o},
year = {2010},
date = {2010-01-01},
journal = {Molecular Simulation},
volume = {36},
number = {13},
pages = {1031--1034},
publisher = {Taylor & Francis},
abstract = {Recently, the encapsulation of β-carotene molecules into carbon nanotubes has been achieved. In this work, we report molecular dynamics simulations and tight-binding density functional-based results for a theoretical study of the encapsulation processes. Our results show that the molecules undergo geometrical deformations when encapsulated with significant changes in their electronic structure. Based on these results, we propose a new interpretation to the changes associated with the β-carotene absorption bands experimentally observed.},
keywords = {Beta-carotene, CNT encapsulation, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
2007
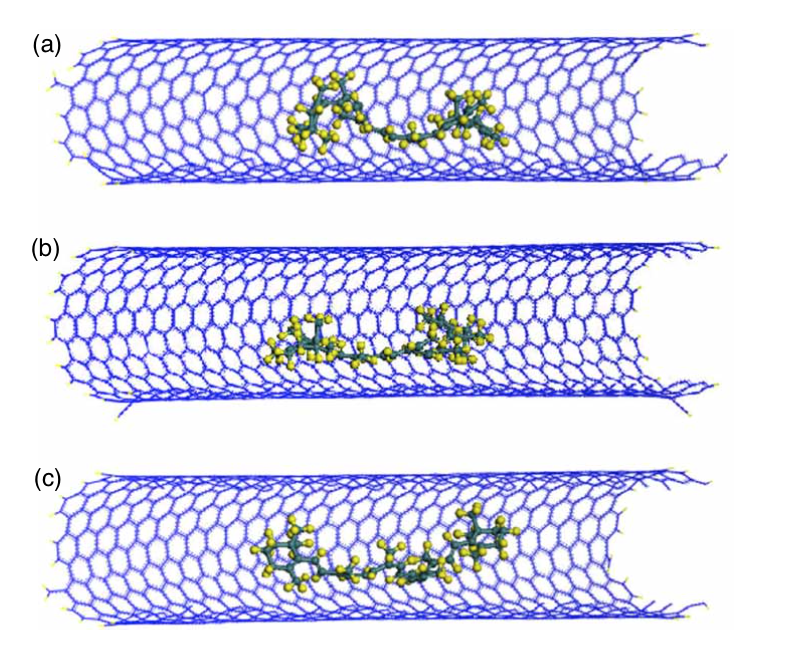
Troche, Karla S; Coluci, Vitor R; Galvao, Douglas S
Atomistic study of the encapsulation of diamondoids inside carbon nanotubes Journal Article
In: arXiv preprint arXiv:0707.1777, 2007.
Abstract | Links | BibTeX | Tags: CNT encapsulation, Diamondoids, Molecular Dynamics
@article{troche2007atomistic,
title = {Atomistic study of the encapsulation of diamondoids inside carbon nanotubes},
author = {Troche, Karla S and Coluci, Vitor R and Galvao, Douglas S},
url = {http://arxiv.org/abs/0707.1777},
year = {2007},
date = {2007-01-01},
journal = {arXiv preprint arXiv:0707.1777},
abstract = {The encapsulation of hydrogen-terminated nanosized diamond fragments (the so-called diamondoids) into armchair single walled carbon nanotubes with diameters in the range of 1.0 up to 2.2 nm has been investigated using classical molecular dynamics simulations. Diameter dependent molecular ordered phases were found for the encapsulation of adamantane (C10H16), diamantane (C14H20), and dihydroxy diamantane (C14H20O2). The same types of chiral ordered phases (double, triple, 4- and 5-stranded helices) observed for the encapsulation of C60 molecules were also observed for diamondoids. On the other hand, some achiral phases comprising layered structures were not observed. Our results also indicate that the modification of diamantane through functionalization with hydroxyl groups can lead to an enhancement of the packing of molecules inside the nanotubes compared to nonfunctionalized compounds. Comparisons to hard-sphere models are also presented revealing differences, specially when more asymmetrical diamondoids are considered. For larger structures (adamantane tetramers) we have not observed long-range ordering for nanotubes with diameters in the range of 1.49 to 2.17 nm but only a tendency to form incomplete helical structures.},
keywords = {CNT encapsulation, Diamondoids, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Azevedo, David L; Sato, Fernando; Galvao, Douglas S; others,
Cobaltocene encapsulation into single-walled carbon nanotubes: A molecular dynamics investigation Journal Article
In: arXiv preprint arXiv:0707.3831, 2007.
Abstract | Links | BibTeX | Tags: CNT encapsulation, Cobaltocene, Molecular Dynamics
@article{azevedo2007cobaltocene,
title = {Cobaltocene encapsulation into single-walled carbon nanotubes: A molecular dynamics investigation},
author = {Azevedo, David L and Sato, Fernando and Galvao, Douglas S and others},
url = {http://arxiv.org/abs/0707.3831},
year = {2007},
date = {2007-01-01},
journal = {arXiv preprint arXiv:0707.3831},
abstract = {Recently (PRL 96, 106804 (2006)) it was suggested that cobaltocene(CC) molecules encapsulated into (7,7) carbon nanotubes (CNT@(7,7)) could be the basis for new spintronic devices. We show here based on impact molecular dynamics and DFT calculations that when dynamical aspects are explicitly considered the CC encapsulation into CNT@(7,7) does not occur, it is prevented by a dynamic barrier mainly due to van der Waals interactions. Our results show that CNT@(13,0) having enough axial space for encapsulation but no enough one to allow freely rotation of the cobaltocene molecule would be a feasible candidate to such application.
},
keywords = {CNT encapsulation, Cobaltocene, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Troche, KS; Coluci, VR; Rurali, R; Galvao, DS
Structural and electronic properties of zigzag carbon nanotubes filled with small fullerenes Journal Article
In: Journal of Physics: Condensed Matter, vol. 19, no. 23, pp. 236222, 2007.
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, CNT encapsulation, Electronic Structure, Fullerenes, Peapods
@article{troche2007structural,
title = {Structural and electronic properties of zigzag carbon nanotubes filled with small fullerenes},
author = {Troche, KS and Coluci, VR and Rurali, R and Galvao, DS},
url = {http://iopscience.iop.org/0953-8984/19/23/236222},
year = {2007},
date = {2007-01-01},
journal = {Journal of Physics: Condensed Matter},
volume = {19},
number = {23},
pages = {236222},
publisher = {IOP Publishing},
abstract = {In this work we investigated the encapsulation of C20 and C30 fullerenes into semiconducting carbon nanotubes to study the possibility of bandgap engineering in such systems. Classical molecular dynamics simulations coupled to tight-binding calculations were used to determine the conformational and electronic properties of carbon nanotubes with an increasing fullerene concentration. We have observed that C20 fullerenes behave similarly to a n-type dopant while C30 can provide p-type doping in some cases. The combined incorporation of both types of fullerenes (hybrid encapsulation) into the same nanotube leads to a behaviour similar to that found in electronic pn-junctions. These aspects can be exploited in the design of nanoelectronic devices using semiconducting carbon nanotubes.
},
keywords = {Carbon Nanotubes, CNT encapsulation, Electronic Structure, Fullerenes, Peapods},
pubstate = {published},
tppubtype = {article}
}
2006
Troche, KS; Coluci, VR; Rurali, R; Galvao, DS
Doping of zigzag carbon nanotubes through the encapsulation of small fullerenes Journal Article
In: arXiv preprint cond-mat/0607197, 2006.
Abstract | Links | BibTeX | Tags: CNT encapsulation, DFT, Molecular Dynamics
@article{troche2006doping,
title = {Doping of zigzag carbon nanotubes through the encapsulation of small fullerenes},
author = {Troche, KS and Coluci, VR and Rurali, R and Galvao, DS},
url = {http://arxiv.org/abs/cond-mat/0607197},
year = {2006},
date = {2006-01-01},
journal = {arXiv preprint cond-mat/0607197},
abstract = {In this work we investigated the encapsulation of C20 and C30 fullerenes into semiconducting carbon nanotubes to study the possibility of bandgap engineering in such systems. Classical molecular dynamics simulations coupled to tight-binding calculations were used to determine the conformational and electronic properties of carbon nanotube supercells containing up to 12 fullerenes. We have observed that C20 fullerenes behave similarly to a p-type dopant while C30 ones work as n-type ones. For larger diameter nanotubes, where fullerene patterns start to differ from the linear arrangements (peapods), the doping features are preserved for both fullerenes, but local disorder plays an important role and significantly alters the electronic structure. The combined incorporation of both fullerene types (hybrid encapsulation) into the same nanotube leads to a behavior similar to that found in electronic junctions in Silicon-based electronic devices. These aspects can be exploited in the design of nanoelectronic devices using semiconducting carbon nanotubes.},
keywords = {CNT encapsulation, DFT, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ

