
Eliezer F; Autreto Oliveira, Pedro AS; Woellner
On the mechanical properties of protomene: A theoretical investigation Journal Article
In: Computational Materials Science, vol. 161, pp. 190-198, 2019.
@article{Oliveira2019c,
title = {On the mechanical properties of protomene: A theoretical investigation},
author = {Oliveira, Eliezer F; Autreto, Pedro AS; Woellner, Cristiano F; Galvao, Douglas S},
year = {2019},
date = {2019-02-07},
journal = {Computational Materials Science},
volume = {161},
pages = {190-198},
abstract = {We report a detailed study through fully atomistic molecular dynamics simulations and DFT calculations on the mechanical properties of protomene. Protomene is a new carbon allotrope composed of a mixture of sp2 and sp3 hybridized states. Our results indicate that protomene presents an anisotropic behavior about tensile deformations. At room temperature, protomene presents an ultimate strength of ~100 GPa and Young's modulus of ~600 GPa, lower than the same for other carbon allotropes. Despite that, protomente presents the highest ultimate strain along the z-direction (~ 24.7%). Our results also show that stretching the protomene along the z-direction or heating it can induce a semiconductor-metallic phase transition, due to a high amount of sp3 bonds that are converted to sp2 ones.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Sanjit; Ozden Bhowmick, Sehmus; Bizão
High temperature quasistatic and dynamic mechanical behavior of interconnected 3D carbon nanotube structures Journal Article
In: Carbon, vol. 142, pp. 291-299, 2019.
@article{Bhowmick2019,
title = {High temperature quasistatic and dynamic mechanical behavior of interconnected 3D carbon nanotube structures},
author = {Bhowmick, Sanjit; Ozden, Sehmus; Bizão, Rafael A; Machado, Leonardo Dantas; Asif, SA Syed; Pugno, Nicola M; Galvao, Douglas S; Tiwary, Chandra Sekhar; Ajayan, PM},
url = {https://www.sciencedirect.com/science/article/pii/S0008622318308911},
doi = {10.1016/j.carbon.2018.09.075},
year = {2019},
date = {2019-02-01},
journal = {Carbon},
volume = {142},
pages = {291-299},
abstract = {Carbon nanotubes (CNTs) are one of the most appealing materials in recent history for both research and commercial interest because of their outstanding physical, chemical, and electrical properties. This is particularly true for 3D arrangements of CNTs which enable their use in larger scale devices and structures. In this paper, the effect of temperature on the quasistatic and dynamic deformation behavior of 3D CNT structures is presented for the first time. An in situ high-temperature nanomechanical instrument was used inside an SEM at high vacuum to investigate mechanical properties of covalently interconnected CNT porous structures in a wide range of temperature. An irreversible bucking at the base of pillar samples was found as a major mode of deformation at room and elevated temperatures. It has been observed that elastic modulus and critical load to first buckle formation decrease progressively with increasing temperature from 25 °C to 750 °C. To understand fatigue resistance, pillars made from this unique structure were compressed to 100 cycles at room temperature and 750 °C. While the structure showed remarkable resistance to fatigue at room temperature, high temperature significantly lowers fatigue resistance. Molecular dynamics (MD) simulation of compression highlights the critical role played by covalent interconnections which prevent localized bending and improve mechanical properties.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
de Sousa, JM; Aguiar, AL; Girao, EC; Fonseca, Alexandre F; AG Filho, Souza; Galvao, Douglas S
Mechanical Properties and Fracture Patterns of Pentagraphene Membranes (under review) Journal Article
In: 2019.
@article{deSousa2019,
title = {Mechanical Properties and Fracture Patterns of Pentagraphene Membranes (under review)},
author = {de Sousa, JM and Aguiar, AL and Girao, EC and Fonseca, Alexandre F and AG Filho, Souza and Galvao, Douglas S},
year = {2019},
date = {2019-01-05},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Fonseca, Alexandre F.; Galvao, Douglas S.
Self-tearing and self-peeling of folded graphene nanoribbons Journal Article
In: Carbon, vol. 143, pp. 230-239, 2019.
@article{Fonseca2019,
title = {Self-tearing and self-peeling of folded graphene nanoribbons},
author = {Alexandre F. Fonseca and Douglas S. Galvao},
url = {https://www.sciencedirect.com/science/article/pii/S0008622318310431},
doi = {10.1016/j.carbon.2018.11.020},
year = {2019},
date = {2019-01-05},
journal = {Carbon},
volume = {143},
pages = {230-239},
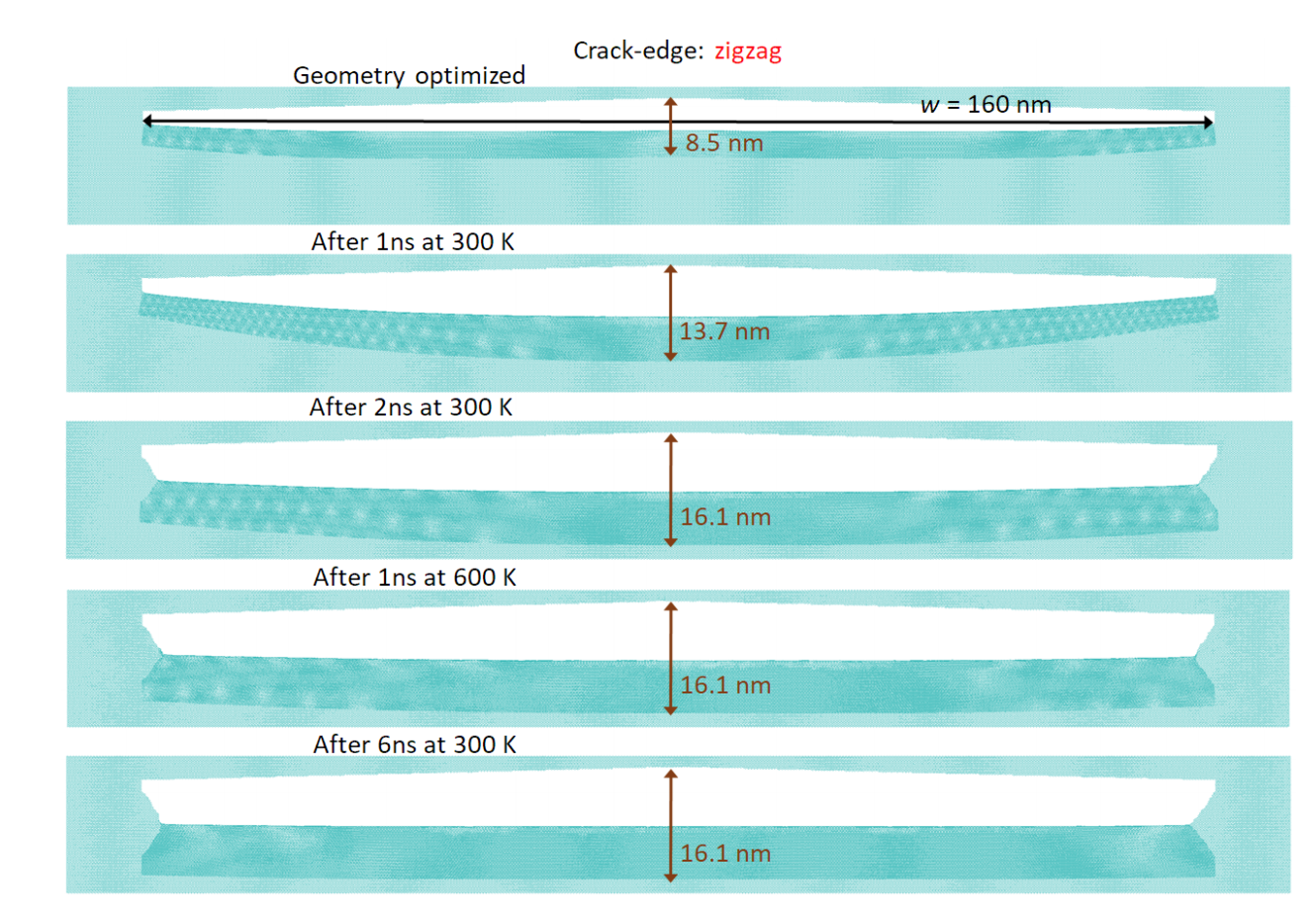
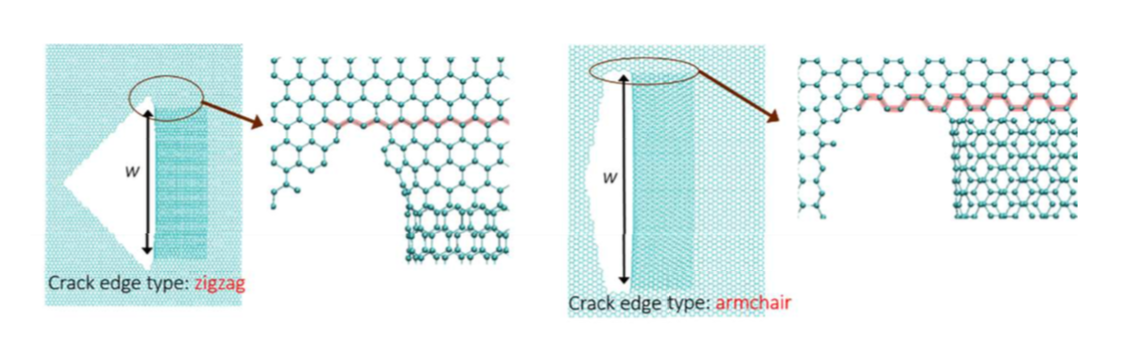
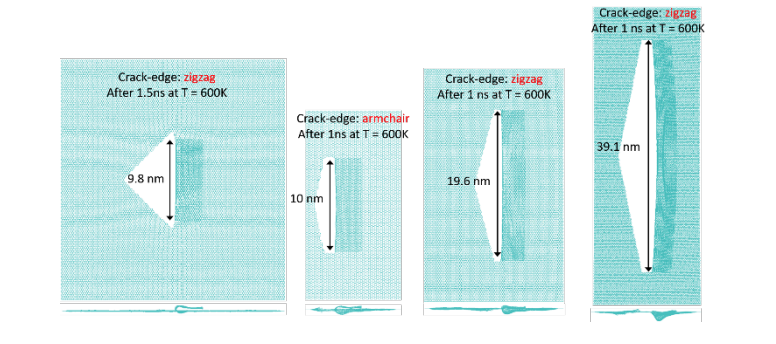
abstract = {A recent experimental study showed that an induced folded flap of graphene can spontaneously drive itself its tearing and peeling off a substrate, thus producing long, micrometer sized, regular trapezoidal-shaped folded graphene nanoribbons. As long as the size of the graphene flaps is above a threshold value, the “tug of war” between the forces of adhesion of graphene-graphene and graphene-substrate, flexural strain of folded region and carbon-carbon (C-C) covalent bonds favor the self-tearing and self-peeling off process. As the detailed information regarding the atomic scale mechanism involved in the process remains not fully understood, we carried out atomistic reactive molecular dynamics simulations to address some features of the process. We show that large thermal fluctuations can prevent the process by increasing the probability of chemical reactions between carbon dangling bonds of adjacent graphene layers. The effects of the strength of attraction between graphene and the substrate on the ribbon growth velocities at the early stages of the phenomenon were also investigated. Structures with initial armchair crack-edges were observed to form more uniform cuts than those having initial zigzag ones. Our results are of importance to help set up new experiments on this phenomenon, especially with samples with nanoscale sized cuts. },
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Eliezer F; Autreto Oliveira, Pedro AS; Woellner
Mechanical Properties of Protomene: A Molecular Dynamics Investigation Journal Article
In: MRS Advances, 2019.
@article{Oliveira2019,
title = {Mechanical Properties of Protomene: A Molecular Dynamics Investigation},
author = {Oliveira, Eliezer F; Autreto, Pedro AS; Woellner, Cristiano F; Galvao, Douglas S},
url = {www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-protomene-a-molecular-dynamics-investigation/CBAC89BDB5942E3353A5C00BD5D0D9CA},
doi = {10.1557/adv.2018.670},
year = {2019},
date = {2019-01-05},
journal = {MRS Advances},
abstract = {Recently, a new class of carbon allotrope called protomene was proposed. This new structure is composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3 carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now, its mechanical properties have not been investigated. In this work, we have investigated protomene mechanical behavior under tensile strain through fully atomistic reactive molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS code. At room temperature, our results show that the protomene is very stable and the obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical fracture.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Pedro AS Autreto Eliezer F Oliveira, Cristiano F Woellner
Mechanical Properties of Protomene: A Molecular Dynamics Investigation Online
2018, (preprint arXiv:1810.09924v1 ).
@online{Oliveira2018g,
title = {Mechanical Properties of Protomene: A Molecular Dynamics Investigation},
author = {Eliezer F Oliveira, Pedro AS Autreto, Cristiano F Woellner, Douglas S Galvao},
url = {https://arxiv.org/abs/1810.09924},
year = {2018},
date = {2018-10-23},
abstract = {Recently, a new class of carbon allotrope called protomene was proposed. This new structure is
composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3
carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations
have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now,
its mechanical properties have not been investigated. In this work, we have investigated
protomene mechanical behavior under tensile strain through fully atomistic reactive
molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS
code. At room temperature, our results show that the protomene is very stable and the
obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest
ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate
strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical
fracture.},
note = {preprint arXiv:1810.09924v1 },
keywords = {},
pubstate = {published},
tppubtype = {online}
}
composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3
carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations
have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now,
its mechanical properties have not been investigated. In this work, we have investigated
protomene mechanical behavior under tensile strain through fully atomistic reactive
molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS
code. At room temperature, our results show that the protomene is very stable and the
obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest
ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate
strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical
fracture.
Alexandre F. Fonseca, Douglas S. Galvao
Self-tearing and self-peeling of folded graphene nanoribbons Online
2018, (preprint arXiv:1808.08872).
@online{Fonseca2018d,
title = { Self-tearing and self-peeling of folded graphene nanoribbons},
author = {Alexandre F. Fonseca, Douglas S. Galvao
},
url = {https://arxiv.org/abs/1808.08872},
year = {2018},
date = {2018-08-27},
abstract = {A recent experimental study showed that an induced folded flap of graphene can spontaneously drive itself its tearing and peeling off a substrate, thus producing long, micrometer sized, regular trapezoidal-shaped folded graphene nanoribbons. As long as the size of the graphene flaps is above a threshold value, the 'tug of war' between the forces of adhesion of graphene-graphene and graphene-substrate, flexural strain of folded region and carbon-carbon (C-C) covalent bonds favor the self-tearing and self-peeling off process. As the detailed information regarding the atomic scale mechanism involved in the process remains not fully understood, we carried out atomistic reactive molecular dynamics simulations to address some features of the process. We show that large thermal fluctuations can prevent the process by increasing the probability of chemical reactions between carbon dangling bonds of adjacent graphene layers. The effects of the strength of attraction between graphene and the substrate on the ribbon growth velocities at the early stages of the phenomenon were also investigated. Structures with initial armchair crack-edges were observed to form more uniform cuts than those having initial zigzag ones. Our results are of importance to help set up new experiments on this phenomenon, especially with samples with nanoscale sized cuts.},
note = {preprint arXiv:1808.08872},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Bizao, Rafael A; Machado, Leonardo D; de Sousa, Jose M; Pugno, Nicola M; Galvao, Douglas S
Scale Effects on the Ballistic Penetration of Graphene Sheets Journal Article
In: Nature Scientific Reports, vol. 8, pp. 6750, 2018.
@article{Bizao2018,
title = {Scale Effects on the Ballistic Penetration of Graphene Sheets},
author = {Bizao, Rafael A and Machado, Leonardo D and de Sousa, Jose M and Pugno, Nicola M and Galvao, Douglas S},
url = {https://www.nature.com/articles/s41598-018-25050-2},
doi = {doi:10.1038/s41598-018-25050-2},
year = {2018},
date = {2018-04-30},
journal = {Nature Scientific Reports},
volume = {8},
pages = {6750},
abstract = {Carbon nanostructures are promising ballistic protection materials, due to their low density and excellent mechanical properties. Recent experimental and computational investigations on the behavior of graphene under impact conditions revealed exceptional energy absorption properties as well. However, the reported numerical and experimental values differ by an order of magnitude. In this work, we combined numerical and analytical modeling to address this issue. In the numerical part, we employed reactive molecular dynamics to carry out ballistic tests on single, double, and triple-layered graphene sheets. We used velocity values within the range tested in experiments. Our numerical and the experimental results were used to determine parameters for a scaling law. We find that the specific penetration energy decreases as the number of layers (N) increases, from ∼15 MJ/kg for N = 1 to ∼0.9 MJ/kg for N = 350, for an impact velocity of 900 m/s. These values are in good agreement with simulations and experiments, within the entire range of N values for which data is presently available. Scale effects explain the apparent discrepancy between simulations and experiments.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Leonardo D Machado Cristiano F Woellner, Pedro AS Autreto; Galvao, Douglas S
Structural Transformations of Carbon and Boron Nitride Nanoscrolls at High Impact Collisions Journal Article
In: Physical Chemistry Chemical Physics, vol. 20, pp. 4911-4916, 2018.
@article{Woellner2018,
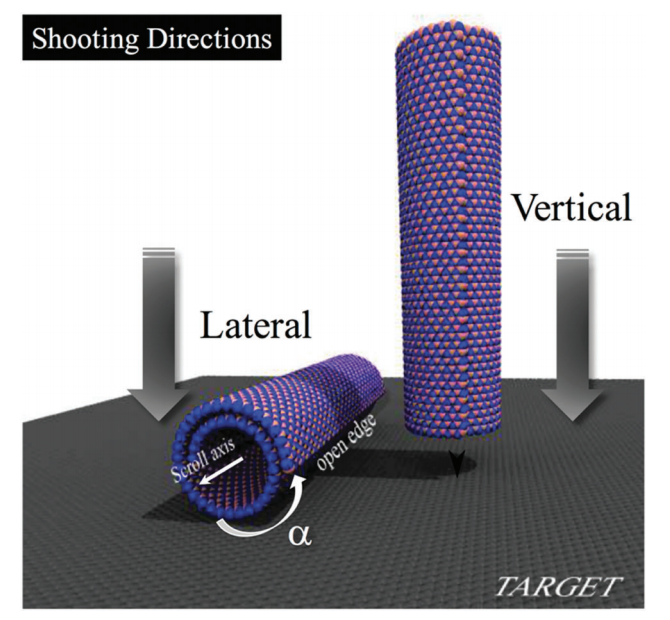
title = {Structural Transformations of Carbon and Boron Nitride Nanoscrolls at High Impact Collisions},
author = {Cristiano F Woellner, Leonardo D Machado, Pedro AS Autreto, Jose M de Sousa, and Douglas S Galvao},
url = {http://pubs.rsc.org/en/content/articlelanding/2018/cp/c7cp07402f#!divAbstract},
doi = {DOI:10.1039/C7CP07402F},
year = {2018},
date = {2018-02-14},
journal = {Physical Chemistry Chemical Physics},
volume = {20},
pages = {4911-4916},
abstract = {The behavior of nanostructures under high strain-rate conditions has been the object of theoretical and
experimental investigations in recent years. For instance, it has been shown that carbon and boron
nitride nanotubes can be unzipped into nanoribbons at high-velocity impacts. However, the response of
many nanostructures to high strain-rate conditions is still unknown. In this work, we have investigated
the mechanical behavior of carbon (CNS) and boron nitride nanoscrolls (BNS) colliding against solid
targets at high velocities, using fully atomistic reactive (ReaxFF) molecular dynamics (MD) simulations.
CNS (BNS) are graphene (boron nitride) membranes rolled up into papyrus-like structures. Their openended
topology leads to unique properties not found in their close-ended analogs, such as nanotubes.
Our results show that collision products are mainly determined by impact velocities and by two
orientation angles, which define the position of the scroll (i) axis and (ii) open edge relative to the target.
Our MD results showed that for appropriate velocities and orientations, large-scale deformations and
nanoscroll fractures could occur. We also observed unscrolling (scrolls going back to quasi-planar
membranes), scroll unzipping into nanoribbons, and significant reconstruction due to breaking and/or
formation of new chemical bonds. For particular edge orientations and velocities, conversion from open
to close-ended topology is also possible, due to the fusion of nanoscroll walls.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
experimental investigations in recent years. For instance, it has been shown that carbon and boron
nitride nanotubes can be unzipped into nanoribbons at high-velocity impacts. However, the response of
many nanostructures to high strain-rate conditions is still unknown. In this work, we have investigated
the mechanical behavior of carbon (CNS) and boron nitride nanoscrolls (BNS) colliding against solid
targets at high velocities, using fully atomistic reactive (ReaxFF) molecular dynamics (MD) simulations.
CNS (BNS) are graphene (boron nitride) membranes rolled up into papyrus-like structures. Their openended
topology leads to unique properties not found in their close-ended analogs, such as nanotubes.
Our results show that collision products are mainly determined by impact velocities and by two
orientation angles, which define the position of the scroll (i) axis and (ii) open edge relative to the target.
Our MD results showed that for appropriate velocities and orientations, large-scale deformations and
nanoscroll fractures could occur. We also observed unscrolling (scrolls going back to quasi-planar
membranes), scroll unzipping into nanoribbons, and significant reconstruction due to breaking and/or
formation of new chemical bonds. For particular edge orientations and velocities, conversion from open
to close-ended topology is also possible, due to the fusion of nanoscroll walls.
Oliveira, Eliezer Fernando; Autreto, Pedro Alves da Silva; Galvao, Douglas Soares
On hardening silver nanocubes by high velocity impacts: a fully atomistic molecular dynamics investigation Journal Article
In: Journal of Materials Science, vol. 53, no. 10, pp. 7486–7492, 2018.
@article{Oliveira2018,
title = {On hardening silver nanocubes by high velocity impacts: a fully atomistic molecular dynamics investigation},
author = {Oliveira, Eliezer Fernando and Autreto, Pedro Alves da Silva and Galvao, Douglas Soares},
url = {https://link.springer.com/article/10.1007/s10853-018-2104-z},
doi = {10.1007/s10853-018-2104-z},
year = {2018},
date = {2018-02-09},
journal = {Journal of Materials Science},
volume = {53},
number = {10},
pages = {7486–7492},
abstract = {Gradient nanograins (GNG) creation in metals has been a promising approach to obtain ultra-strong materials. Recently, R. Thevamaran et al. (Science 354:312 in 2016) proposed a single-step method based on high-velocity impacts of silver nanocubes (SNC) to produce almost perfect GNG. However, after certain time, these grains spontaneously coalesce, which compromises the induced hardening and other mechanical properties. To better understand these processes, a detailed investigation at the atomic scale of the deformation/hardening mechanisms are needed, which is one of the objectives of the present work. We carried out fully atomistic molecular dynamics (MD) simulations of silver nanocubes at high impact velocity values using realistic structural models. Our MD results suggest that besides the GNG mechanisms, the observed SNC hardening could be also the result of the existence of polycrystalline arrangements formed by HCP domains encapsulated by FCC ones in the smashed SNC. This can be a new way to design ultra-strong materials, even in the absence of GNG domains.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
de Sousa, J. M.; Aguiar, A. L.; Girao, E. C.; Fonseca, Alexandre F.; Filho, A. G. Souza; Galvao, Douglas S.
Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 97-102, 2018.
@article{deSousa2018b,
title = {Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study},
author = {J. M. de Sousa and A. L. Aguiar and E. C. Girao and Alexandre F. Fonseca and A. G. Souza Filho and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-pentagraphenebased-nanotubes-a-molecular-dynamics-study/289AB70DADF20059BB8FCC9EF07B97AB},
doi = { https://doi.org/10.1557/adv.2018.160},
year = {2018},
date = {2018-02-06},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = {97-102},
abstract = {The study of the mechanical properties of nanostructured systems has gained importance in theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of the strongest nanomaterials found in nature, with Young’s Modulus (EY) in the order 1.25 TPa. One interesting question is about the possibility of generating new nanostructures with 1D symmetry and with similar and/or superior CNT properties. In this work, we present a study on the dynamical, structural, mechanical properties, fracture patterns and EY values for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized states) in the same form that CNTs are formed from rolling up graphene membranes. We carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We have considered zigzag-like and armchair-like PGNTs of different diameters. Our results show that PGNTs present EY ∼ 800 GPa with distinct elastic behavior in relation to CNTs, mainly associated with mechanical failure, chirality dependent fracture patterns and extensive structural reconstructions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Fonseca, Alexandre F.; Galvao, Douglas S.
Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation Journal Article
In: MRS Advances, vol. 3, no. 8-9, pp. 460-465, 2018.
@article{Fonseca2018,
title = {Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation},
author = {Alexandre F. Fonseca and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/selfdriven-graphene-tearing-and-peeling-a-fully-atomistic-molecular-dynamics-investigation/BFC76FC4479AA617E16FA6AC7AB4D487},
doi = {https://doi.org/10.1557/adv.2018.120},
year = {2018},
date = {2018-01-30},
journal = {MRS Advances},
volume = {3},
number = {8-9},
pages = {460-465},
abstract = {In spite of years of intense research, graphene continues to produce surprising results. Recently, it was experimentally observed that under certain conditions graphene can self-drive its tearing and peeling from substrates. This process can generate long, micrometer sized, folded nanoribbons without the action of any external forces. Also, during this cracking-like propagation process, the width of the graphene folded ribbon continuously decreases and the process only stops when the width reaches about few hundreds nanometers in size. It is believed that interplay between the strain energy of folded regions, breaking of carbon-carbon covalent bonds, and adhesion of graphene-graphene and graphene-substrate are the most fundamental features of this process, although the detailed mechanisms at atomic scale remain unclear. In order to gain further insights on these processes we carried out fully atomistic reactive molecular dynamics simulations using the AIREBO potential as available in the LAMMPS computational package. Although the reported tearing/peeling experimental observations were only to micrometer sized structures, our results showed that they could also occur at nanometer scale. Our preliminary results suggest that the graphene tearing/peeling process originates from thermal energy fluctuations that results in broken bonds, followed by strain release that creates a local elastic wave that can either reinforce the process, similar to a whip cracking propagation, or undermine it by producing carbon dangling bonds that evolve to the formation of bonds between the two layers of graphene. As the process continues in time and the folded graphene decreases in width, the carbon-carbon bonds at the ribbon edge and interlayer bonds get less stressed, thermal fluctuations become unable to break them and the process stops.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Azevedo, David L.; Bizao, Rafael A.; Galvao, Douglas S.
Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets Online
2018, (preprint arXiv:1801.05346).
@online{Azevedo2018b,
title = {Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets},
author = {David L. Azevedo and Rafael A. Bizao and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.05346},
year = {2018},
date = {2018-01-18},
abstract = {The superior mechanical properties and low density of carbon nanostructures make them promising ballistic protection materials, stimulating investigations on their high-strain-rate behavior. Recent experiments and simulations revealed graphene possesses exceptional energy absorption properties. In this work, we analyzed through fully atomistic molecular dynamics simulations the ballistic performance of a carbon-based material recently proposed named penta-graphene. Our results show that the fracture pattern is more spherical (no petals formation like observed for graphene). The estimated penetration energy for pentagraphene structures considered here was of 37.69 MJ/Kg, far superior to graphene (29.8 MJ/Kg) under same conditions. These preliminary results are suggestive that pentagraphene could be an excellent material for ballistic applications.},
note = {preprint arXiv:1801.05346},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Fonseca, Alexandre F.; Galvao, Douglas S.
Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation Online
2018, (preprint arXiv:1801.05354).
@online{Fonseca2018b,
title = {Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation},
author = {Alexandre F. Fonseca and Douglas S. Galvao
},
url = {https://arxiv.org/abs/1801.05354},
year = {2018},
date = {2018-01-17},
abstract = {In spite of years of intense research, graphene continues to produce surprising results. Recently, it was experimentally observed that under certain conditions graphene can self-drive its tearing and peeling from substrates. This process can generate long, micrometer sized, folded nanoribbons without the action of any external forces. Also, during this cracking-like propagation process, the width of the graphene folded ribbon continuously decreases and the process only stops when the width reaches about few hundreds nanometers in size. It is believed that interplay between the strain energy of folded regions, breaking of carbon-carbon covalent bonds, and adhesion of graphene-graphene and graphene-substrate are the most fundamental features of this process, although the detailed mechanisms at atomic scale remain unclear. In order to gain further insights on these processes we carried out fully atomistic reactive molecular dynamics simulations using the AIREBO potential as available in the LAMMPS computational package. Although the reported tearing/peeling experimental observations were only to micrometer sized structures, our results showed that they could also occur at nanometer scale. Our preliminary results suggest that the graphene tearing/peeling process originates from thermal energy fluctuations that results in broken bonds, followed by strain release that creates a local elastic wave that can either reinforce the process, similar to a whip cracking propagation, or undermine it by producing carbon dangling bonds that evolve to the formation of bonds between the two layers of graphene. As the process continues in time and the folded graphene decreases in width, the carbon-carbon bonds at the ribbon edge and interlayer bonds get less stressed, thermal fluctuations become unable to break them and the process stops.},
note = {preprint arXiv:1801.05354},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
de Sousa, J. M.; Aguiar, A. L.; Girao, E. C.; Fonseca, Alexandre F.; Filho, A. G. Souza; Galvao, Douglas S.
Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 67-72, 2018.
@article{deSousa2018c,
title = {Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation},
author = {J. M. de Sousa and A. L. Aguiar and E. C. Girao and Alexandre F. Fonseca and A. G. Souza Filho and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-phagraphene-membranes-a-fully-atomistic-molecular-dynamics-investigation/3ADC3F3B0052AB6632E8681404948E7B},
doi = {DOI: 10.1557/adv.2018. 54},
year = {2018},
date = {2018-01-15},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = {67-72},
abstract = {Recently, a new 2D carbon allotrope structure, named phagraphene (PG), was proposed. PG has a densely array of penta-hexa-hepta-graphene carbon rings. PG was shown to present low and anisotropic thermal conductivity and it is believed that this anisotropy should be also reflected in its mechanical properties. Although PG mechanical properties have been investigated, a detailed and comprehensive study is still lacking. In the present work we have carried out fully atomistic reactive molecular dynamics simulations using the ReaxFF force field, to investigate the mechanical properties and fracture patterns of PG membranes. The Young's modulus values of the PG membranes were estimated from the stress-strain curves. Our results show that these curves present three distinct regimes: one regime where ripples dominate the structure and mechanical properties of the PG membranes; an elastic regime where the membranes exhibit fully planar configurations; and finally am inelastic regime where permanent deformations happened to the PG membrane up to the mechanical failure or fracture.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
de Sousa, J. M.; Aguiar, A. L.; Girao, E. C.; Fonseca, Alexandre F.; Filho, A. G. Sousa; Galvao, Douglas S.
Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study Online
2018, (preprint arXiv:1801.04269).
@online{deSousa2018d,
title = {Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study},
author = {J. M. de Sousa and A. L. Aguiar and E. C. Girao and Alexandre F. Fonseca and A. G. Sousa Filho and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.04269},
year = {2018},
date = {2018-01-15},
abstract = {The study of the mechanical properties of nanostructured systems has gained importance in
theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of
the strongest nanomaterials found in nature, with Young's Modulus (YM) in the order 1.25
TPa. One interesting question is about the possibility of generating new nanostructures with
1D symmetry and with similar and/or superior CNT properties. In this work, we present a
study on the dynamical, structural, mechanical properties, fracture patterns and YM values
for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These
tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional
structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized
states) in the same form that CNTs are formed from rolling up graphene membranes. We
carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We
have considered zigzag-like and armchair-like PGNTs of different diameters. Our results
show that PGNTs present YM ~ 800 GPa with distinct elastic behavior in relation to CNTs,
mainly associated with mechanical failure, chirality dependent fracture patterns and extensive
structural reconstructions},
note = {preprint arXiv:1801.04269},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of
the strongest nanomaterials found in nature, with Young's Modulus (YM) in the order 1.25
TPa. One interesting question is about the possibility of generating new nanostructures with
1D symmetry and with similar and/or superior CNT properties. In this work, we present a
study on the dynamical, structural, mechanical properties, fracture patterns and YM values
for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These
tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional
structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized
states) in the same form that CNTs are formed from rolling up graphene membranes. We
carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We
have considered zigzag-like and armchair-like PGNTs of different diameters. Our results
show that PGNTs present YM ~ 800 GPa with distinct elastic behavior in relation to CNTs,
mainly associated with mechanical failure, chirality dependent fracture patterns and extensive
structural reconstructions
de Sousa, Jose M.; Aguiar, Acrisio L.; Girao, Eduardo C.; Fonseca, Alexandre F.; Antonio G. Sousa Filho,; Galvao, Douglas S.
Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation Online
2018, (preprint arXiv:1801.04292).
@online{deSousa2018e,
title = {Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Jose M. de Sousa and Acrisio L. Aguiar and Eduardo C. Girao and Alexandre F. Fonseca and Antonio G. Sousa Filho, and Douglas S. Galvao
},
url = {https://arxiv.org/abs/1801.04292},
year = {2018},
date = {2018-01-12},
abstract = {Recently, a new 2D carbon allotrope structure, named phagraphene (PG), was proposed. PG has a densely array of penta-hexa-hepta-graphene carbon rings. PG was shown to present low and anisotropic thermal conductivity and it is believed that this anisotropy should be also reflected in its mechanical properties. Although PG mechanical properties have been investigated, a detailed and comprehensive study is still lacking. In the present work we have carried out fully atomistic reactive molecular dynamics simulations using the ReaxFF force field, to investigate the mechanical properties and fracture patterns of PG membranes. The Young's modulus values of the PG membranes were estimated from the stress-strain curves. Our results show that these curves present three distinct regimes: one regime where ripples dominate the structure and mechanical properties of the PG membranes; an elastic regime where the membranes exhibit fully planar configurations; and finally a plastic regime where permanent deformations happened to the PG membrane up to the mechanical failure or fracture.},
note = {preprint arXiv:1801.04292},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
de Sousa, Jose M.; Aguiar, Acrisio L.; Girao, Eduardo C.; Fonseca, Alexandre F.; Antonio G. Souza Filho,; Galvao, Douglas S.
Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study Online
2018, (preprint arXiv:1801.04269).
@online{deSousa2018f,
title = {Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study},
author = {Jose M. de Sousa and Acrisio L. Aguiar and Eduardo C. Girao and Alexandre F. Fonseca and Antonio G. Souza Filho, and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.04269},
year = {2018},
date = {2018-01-12},
abstract = {The study of the mechanical properties of nanostructured systems has gained importance in theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of the strongest nanomaterials found in nature, with Young's Modulus (YM) in the order 1.25 TPa. One interesting question is about the possibility of generating new nanostructures with 1D symmetry and with similar and/or superior CNT properties. In this work, we present a study on the dynamical, structural, mechanical properties, fracture patterns and YM values for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized states) in the same form that CNTs are formed from rolling up graphene membranes. We carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We have considered zigzag-like and armchair-like PGNTs of different diameters. Our results show that PGNTs present YM ~ 800 GPa with distinct elastic behavior in relation to CNTs, mainly associated with mechanical failure, chirality dependent fracture patterns and extensive structural reconstructions.},
note = {preprint arXiv:1801.04269},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Azevedo, David L.; Bizao, Rafael A.; Galvao, Douglas S.
Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets Journal Article
In: MRS Advances, vol. 3, no. 8-9, pp. 431-435, 2018.
@article{Azevedo2018,
title = {Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets},
author = {David L. Azevedo and Rafael A. Bizao and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/molecular-dynamics-simulations-of-ballistic-penetration-of-pentagraphene-sheets/8759C0815840EDE83896EF4A17278228},
doi = {https://doi.org/10.1557/adv.2018.61},
year = {2018},
date = {2018-01-06},
journal = {MRS Advances},
volume = {3},
number = {8-9},
pages = {431-435},
abstract = {The search for new materials with low density and superior mechanical properties is a very intense and stimulating investigation area. These new materials could provide potential application for ballistic protection. Recent experiments and simulations revealed graphene possesses exceptional energy absorption properties. In this work, we analysed through fully atomistic molecular dynamics simulations the ballistic performance of a carbon-based material recently proposed named penta-graphene. Our results show that the fracture pattern is more spherical (no petals formation like observed for graphene). The estimated penetration energy for single-layer penta-graphene structures obtained here was d_1penta∼37.7 MJ/kg, and is comparable with recently results obtained for graphene: d_(1graphene)∼29.0 MJ/kg and d_(1graphene)∼40.8 MJ/kg under similar conditions. These preliminary results are suggestive that penta-graphene could be an excellent material for ballistic applications.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Oliveira, Eliezer Fernando; da Silva Autreto, Pedro Alves; Galvao, Douglas Soares
Silver Hardening via Hypersonic Impacts Journal Article
In: MRS Advances, vol. 3, no. 8-9, pp. 489-494, 2018.
@article{Oliveira2018b,
title = {Silver Hardening via Hypersonic Impacts},
author = {Eliezer Fernando Oliveira and Pedro Alves da Silva Autreto and Douglas Soares Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/silver-hardening-via-hypersonic-impacts/6A35FAB117B4FD244BBD11A64CD25160},
doi = {DOI: 10.1557/adv.2018. 173},
year = {2018},
date = {2018-01-01},
journal = {MRS Advances},
volume = {3},
number = {8-9},
pages = {489-494},
abstract = {The search for new ultra strong materials has been a very active research area. With relation to metals, a successful way to improve their strength is by the creation of a gradient of nanograins (GNG) inside the material. Recently, R. Thevamaran et al. [Science v354, 312- 316 (2016)] propose a single step method based on high velocity impact of silver nanocubes to produce high-quality GNG. This method consists of producing high impact collisions of silver cubes at hypersonic velocity (~400 m/s) against a rigid wall. Although they observed an improvement in the mechanical properties of the silver after the impact, the GNG creation and the strengthening mechanism at nanoscale remain unclear. In order to gain further insights about these mechanisms, we carried out fully atomistic molecular dynamics simulations (MD) to investigate the atomic conformations/rearrangements during and after high impact collisions of silver nanocubes at ultrasonic velocity. Our results indicate the co- existence of polycrystalline arrangements after the impact formed by core HCP domains surrounded by FCC ones, which could also contribute to explain the structural hardening.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2019
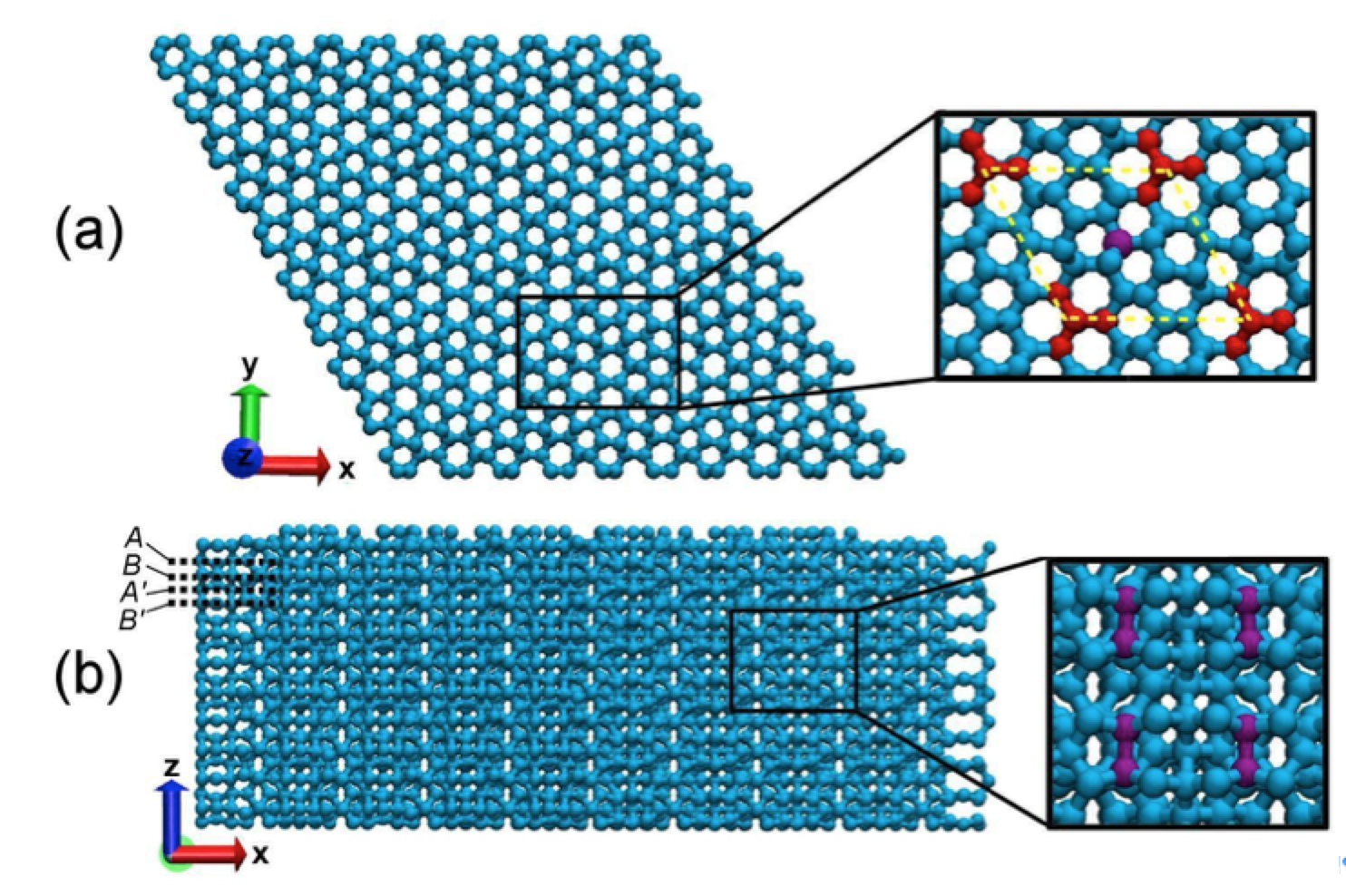
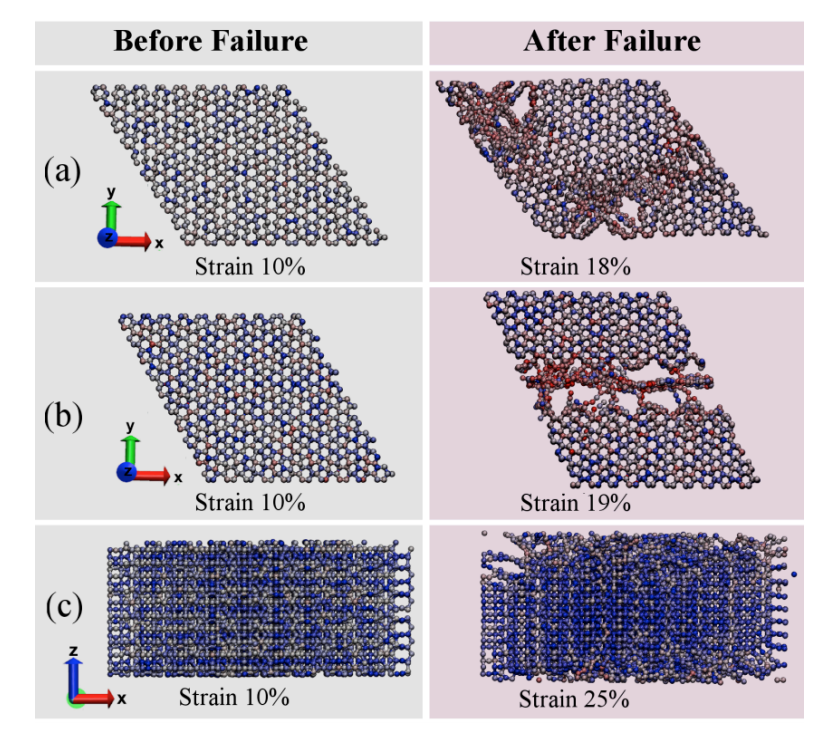
Eliezer F; Autreto Oliveira, Pedro AS; Woellner
On the mechanical properties of protomene: A theoretical investigation Journal Article
In: Computational Materials Science, vol. 161, pp. 190-198, 2019.
Abstract | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, protomene
@article{Oliveira2019c,
title = {On the mechanical properties of protomene: A theoretical investigation},
author = {Oliveira, Eliezer F; Autreto, Pedro AS; Woellner, Cristiano F; Galvao, Douglas S},
year = {2019},
date = {2019-02-07},
journal = {Computational Materials Science},
volume = {161},
pages = {190-198},
abstract = {We report a detailed study through fully atomistic molecular dynamics simulations and DFT calculations on the mechanical properties of protomene. Protomene is a new carbon allotrope composed of a mixture of sp2 and sp3 hybridized states. Our results indicate that protomene presents an anisotropic behavior about tensile deformations. At room temperature, protomene presents an ultimate strength of ~100 GPa and Young's modulus of ~600 GPa, lower than the same for other carbon allotropes. Despite that, protomente presents the highest ultimate strain along the z-direction (~ 24.7%). Our results also show that stretching the protomene along the z-direction or heating it can induce a semiconductor-metallic phase transition, due to a high amount of sp3 bonds that are converted to sp2 ones.},
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, protomene},
pubstate = {published},
tppubtype = {article}
}
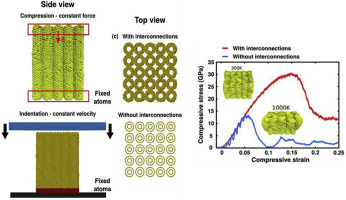
Sanjit; Ozden Bhowmick, Sehmus; Bizão
High temperature quasistatic and dynamic mechanical behavior of interconnected 3D carbon nanotube structures Journal Article
In: Carbon, vol. 142, pp. 291-299, 2019.
Abstract | Links | BibTeX | Tags: CNT, Fracture, Mechanical Properties, Molecular Dynamics
@article{Bhowmick2019,
title = {High temperature quasistatic and dynamic mechanical behavior of interconnected 3D carbon nanotube structures},
author = {Bhowmick, Sanjit; Ozden, Sehmus; Bizão, Rafael A; Machado, Leonardo Dantas; Asif, SA Syed; Pugno, Nicola M; Galvao, Douglas S; Tiwary, Chandra Sekhar; Ajayan, PM},
url = {https://www.sciencedirect.com/science/article/pii/S0008622318308911},
doi = {10.1016/j.carbon.2018.09.075},
year = {2019},
date = {2019-02-01},
journal = {Carbon},
volume = {142},
pages = {291-299},
abstract = {Carbon nanotubes (CNTs) are one of the most appealing materials in recent history for both research and commercial interest because of their outstanding physical, chemical, and electrical properties. This is particularly true for 3D arrangements of CNTs which enable their use in larger scale devices and structures. In this paper, the effect of temperature on the quasistatic and dynamic deformation behavior of 3D CNT structures is presented for the first time. An in situ high-temperature nanomechanical instrument was used inside an SEM at high vacuum to investigate mechanical properties of covalently interconnected CNT porous structures in a wide range of temperature. An irreversible bucking at the base of pillar samples was found as a major mode of deformation at room and elevated temperatures. It has been observed that elastic modulus and critical load to first buckle formation decrease progressively with increasing temperature from 25 °C to 750 °C. To understand fatigue resistance, pillars made from this unique structure were compressed to 100 cycles at room temperature and 750 °C. While the structure showed remarkable resistance to fatigue at room temperature, high temperature significantly lowers fatigue resistance. Molecular dynamics (MD) simulation of compression highlights the critical role played by covalent interconnections which prevent localized bending and improve mechanical properties.},
keywords = {CNT, Fracture, Mechanical Properties, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
de Sousa, JM; Aguiar, AL; Girao, EC; Fonseca, Alexandre F; AG Filho, Souza; Galvao, Douglas S
Mechanical Properties and Fracture Patterns of Pentagraphene Membranes (under review) Journal Article
In: 2019.
BibTeX | Tags: Fracture, Molecular Dynamics, pentagraphene
@article{deSousa2019,
title = {Mechanical Properties and Fracture Patterns of Pentagraphene Membranes (under review)},
author = {de Sousa, JM and Aguiar, AL and Girao, EC and Fonseca, Alexandre F and AG Filho, Souza and Galvao, Douglas S},
year = {2019},
date = {2019-01-05},
keywords = {Fracture, Molecular Dynamics, pentagraphene},
pubstate = {published},
tppubtype = {article}
}

Fonseca, Alexandre F.; Galvao, Douglas S.
Self-tearing and self-peeling of folded graphene nanoribbons Journal Article
In: Carbon, vol. 143, pp. 230-239, 2019.
Abstract | Links | BibTeX | Tags: Fracture, Graphene, Mechanical Properties, Molecular Dynamics
@article{Fonseca2019,
title = {Self-tearing and self-peeling of folded graphene nanoribbons},
author = {Alexandre F. Fonseca and Douglas S. Galvao},
url = {https://www.sciencedirect.com/science/article/pii/S0008622318310431},
doi = {10.1016/j.carbon.2018.11.020},
year = {2019},
date = {2019-01-05},
journal = {Carbon},
volume = {143},
pages = {230-239},
abstract = {A recent experimental study showed that an induced folded flap of graphene can spontaneously drive itself its tearing and peeling off a substrate, thus producing long, micrometer sized, regular trapezoidal-shaped folded graphene nanoribbons. As long as the size of the graphene flaps is above a threshold value, the “tug of war” between the forces of adhesion of graphene-graphene and graphene-substrate, flexural strain of folded region and carbon-carbon (C-C) covalent bonds favor the self-tearing and self-peeling off process. As the detailed information regarding the atomic scale mechanism involved in the process remains not fully understood, we carried out atomistic reactive molecular dynamics simulations to address some features of the process. We show that large thermal fluctuations can prevent the process by increasing the probability of chemical reactions between carbon dangling bonds of adjacent graphene layers. The effects of the strength of attraction between graphene and the substrate on the ribbon growth velocities at the early stages of the phenomenon were also investigated. Structures with initial armchair crack-edges were observed to form more uniform cuts than those having initial zigzag ones. Our results are of importance to help set up new experiments on this phenomenon, especially with samples with nanoscale sized cuts. },
keywords = {Fracture, Graphene, Mechanical Properties, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
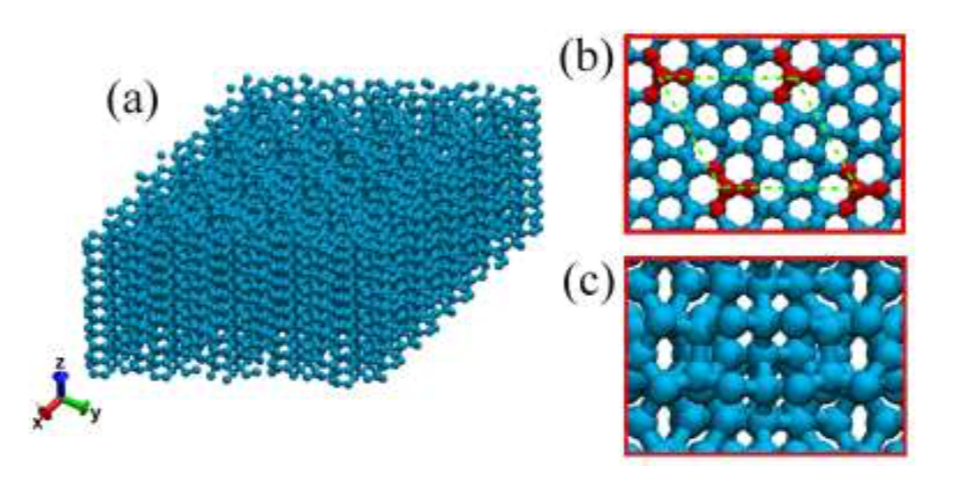
Eliezer F; Autreto Oliveira, Pedro AS; Woellner
Mechanical Properties of Protomene: A Molecular Dynamics Investigation Journal Article
In: MRS Advances, 2019.
Abstract | Links | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, protomene
@article{Oliveira2019,
title = {Mechanical Properties of Protomene: A Molecular Dynamics Investigation},
author = {Oliveira, Eliezer F; Autreto, Pedro AS; Woellner, Cristiano F; Galvao, Douglas S},
url = {www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-protomene-a-molecular-dynamics-investigation/CBAC89BDB5942E3353A5C00BD5D0D9CA},
doi = {10.1557/adv.2018.670},
year = {2019},
date = {2019-01-05},
journal = {MRS Advances},
abstract = {Recently, a new class of carbon allotrope called protomene was proposed. This new structure is composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3 carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now, its mechanical properties have not been investigated. In this work, we have investigated protomene mechanical behavior under tensile strain through fully atomistic reactive molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS code. At room temperature, our results show that the protomene is very stable and the obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical fracture.
},
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, protomene},
pubstate = {published},
tppubtype = {article}
}
2018
Pedro AS Autreto Eliezer F Oliveira, Cristiano F Woellner
Mechanical Properties of Protomene: A Molecular Dynamics Investigation Online
2018, (preprint arXiv:1810.09924v1 ).
Abstract | Links | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, protomene
@online{Oliveira2018g,
title = {Mechanical Properties of Protomene: A Molecular Dynamics Investigation},
author = {Eliezer F Oliveira, Pedro AS Autreto, Cristiano F Woellner, Douglas S Galvao},
url = {https://arxiv.org/abs/1810.09924},
year = {2018},
date = {2018-10-23},
abstract = {Recently, a new class of carbon allotrope called protomene was proposed. This new structure is
composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3
carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations
have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now,
its mechanical properties have not been investigated. In this work, we have investigated
protomene mechanical behavior under tensile strain through fully atomistic reactive
molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS
code. At room temperature, our results show that the protomene is very stable and the
obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest
ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate
strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical
fracture.},
note = {preprint arXiv:1810.09924v1 },
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, protomene},
pubstate = {published},
tppubtype = {online}
}
composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3
carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations
have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now,
its mechanical properties have not been investigated. In this work, we have investigated
protomene mechanical behavior under tensile strain through fully atomistic reactive
molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS
code. At room temperature, our results show that the protomene is very stable and the
obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest
ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate
strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical
fracture.
Alexandre F. Fonseca, Douglas S. Galvao
Self-tearing and self-peeling of folded graphene nanoribbons Online
2018, (preprint arXiv:1808.08872).
Abstract | Links | BibTeX | Tags: Fracture, graphene nanoribbons, Mechanical Properties, Molecular Dynamics
@online{Fonseca2018d,
title = { Self-tearing and self-peeling of folded graphene nanoribbons},
author = {Alexandre F. Fonseca, Douglas S. Galvao
},
url = {https://arxiv.org/abs/1808.08872},
year = {2018},
date = {2018-08-27},
abstract = {A recent experimental study showed that an induced folded flap of graphene can spontaneously drive itself its tearing and peeling off a substrate, thus producing long, micrometer sized, regular trapezoidal-shaped folded graphene nanoribbons. As long as the size of the graphene flaps is above a threshold value, the 'tug of war' between the forces of adhesion of graphene-graphene and graphene-substrate, flexural strain of folded region and carbon-carbon (C-C) covalent bonds favor the self-tearing and self-peeling off process. As the detailed information regarding the atomic scale mechanism involved in the process remains not fully understood, we carried out atomistic reactive molecular dynamics simulations to address some features of the process. We show that large thermal fluctuations can prevent the process by increasing the probability of chemical reactions between carbon dangling bonds of adjacent graphene layers. The effects of the strength of attraction between graphene and the substrate on the ribbon growth velocities at the early stages of the phenomenon were also investigated. Structures with initial armchair crack-edges were observed to form more uniform cuts than those having initial zigzag ones. Our results are of importance to help set up new experiments on this phenomenon, especially with samples with nanoscale sized cuts.},
note = {preprint arXiv:1808.08872},
keywords = {Fracture, graphene nanoribbons, Mechanical Properties, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}
Bizao, Rafael A; Machado, Leonardo D; de Sousa, Jose M; Pugno, Nicola M; Galvao, Douglas S
Scale Effects on the Ballistic Penetration of Graphene Sheets Journal Article
In: Nature Scientific Reports, vol. 8, pp. 6750, 2018.
Abstract | Links | BibTeX | Tags: Fracture, Graphene, impact, Molecular Dynamics
@article{Bizao2018,
title = {Scale Effects on the Ballistic Penetration of Graphene Sheets},
author = {Bizao, Rafael A and Machado, Leonardo D and de Sousa, Jose M and Pugno, Nicola M and Galvao, Douglas S},
url = {https://www.nature.com/articles/s41598-018-25050-2},
doi = {doi:10.1038/s41598-018-25050-2},
year = {2018},
date = {2018-04-30},
journal = {Nature Scientific Reports},
volume = {8},
pages = {6750},
abstract = {Carbon nanostructures are promising ballistic protection materials, due to their low density and excellent mechanical properties. Recent experimental and computational investigations on the behavior of graphene under impact conditions revealed exceptional energy absorption properties as well. However, the reported numerical and experimental values differ by an order of magnitude. In this work, we combined numerical and analytical modeling to address this issue. In the numerical part, we employed reactive molecular dynamics to carry out ballistic tests on single, double, and triple-layered graphene sheets. We used velocity values within the range tested in experiments. Our numerical and the experimental results were used to determine parameters for a scaling law. We find that the specific penetration energy decreases as the number of layers (N) increases, from ∼15 MJ/kg for N = 1 to ∼0.9 MJ/kg for N = 350, for an impact velocity of 900 m/s. These values are in good agreement with simulations and experiments, within the entire range of N values for which data is presently available. Scale effects explain the apparent discrepancy between simulations and experiments.},
keywords = {Fracture, Graphene, impact, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Leonardo D Machado Cristiano F Woellner, Pedro AS Autreto; Galvao, Douglas S
Structural Transformations of Carbon and Boron Nitride Nanoscrolls at High Impact Collisions Journal Article
In: Physical Chemistry Chemical Physics, vol. 20, pp. 4911-4916, 2018.
Abstract | Links | BibTeX | Tags: Fracture, impact, Molecular Dynamics, scroll
@article{Woellner2018,
title = {Structural Transformations of Carbon and Boron Nitride Nanoscrolls at High Impact Collisions},
author = {Cristiano F Woellner, Leonardo D Machado, Pedro AS Autreto, Jose M de Sousa, and Douglas S Galvao},
url = {http://pubs.rsc.org/en/content/articlelanding/2018/cp/c7cp07402f#!divAbstract},
doi = {DOI:10.1039/C7CP07402F},
year = {2018},
date = {2018-02-14},
journal = {Physical Chemistry Chemical Physics},
volume = {20},
pages = {4911-4916},
abstract = {The behavior of nanostructures under high strain-rate conditions has been the object of theoretical and
experimental investigations in recent years. For instance, it has been shown that carbon and boron
nitride nanotubes can be unzipped into nanoribbons at high-velocity impacts. However, the response of
many nanostructures to high strain-rate conditions is still unknown. In this work, we have investigated
the mechanical behavior of carbon (CNS) and boron nitride nanoscrolls (BNS) colliding against solid
targets at high velocities, using fully atomistic reactive (ReaxFF) molecular dynamics (MD) simulations.
CNS (BNS) are graphene (boron nitride) membranes rolled up into papyrus-like structures. Their openended
topology leads to unique properties not found in their close-ended analogs, such as nanotubes.
Our results show that collision products are mainly determined by impact velocities and by two
orientation angles, which define the position of the scroll (i) axis and (ii) open edge relative to the target.
Our MD results showed that for appropriate velocities and orientations, large-scale deformations and
nanoscroll fractures could occur. We also observed unscrolling (scrolls going back to quasi-planar
membranes), scroll unzipping into nanoribbons, and significant reconstruction due to breaking and/or
formation of new chemical bonds. For particular edge orientations and velocities, conversion from open
to close-ended topology is also possible, due to the fusion of nanoscroll walls.},
keywords = {Fracture, impact, Molecular Dynamics, scroll},
pubstate = {published},
tppubtype = {article}
}
experimental investigations in recent years. For instance, it has been shown that carbon and boron
nitride nanotubes can be unzipped into nanoribbons at high-velocity impacts. However, the response of
many nanostructures to high strain-rate conditions is still unknown. In this work, we have investigated
the mechanical behavior of carbon (CNS) and boron nitride nanoscrolls (BNS) colliding against solid
targets at high velocities, using fully atomistic reactive (ReaxFF) molecular dynamics (MD) simulations.
CNS (BNS) are graphene (boron nitride) membranes rolled up into papyrus-like structures. Their openended
topology leads to unique properties not found in their close-ended analogs, such as nanotubes.
Our results show that collision products are mainly determined by impact velocities and by two
orientation angles, which define the position of the scroll (i) axis and (ii) open edge relative to the target.
Our MD results showed that for appropriate velocities and orientations, large-scale deformations and
nanoscroll fractures could occur. We also observed unscrolling (scrolls going back to quasi-planar
membranes), scroll unzipping into nanoribbons, and significant reconstruction due to breaking and/or
formation of new chemical bonds. For particular edge orientations and velocities, conversion from open
to close-ended topology is also possible, due to the fusion of nanoscroll walls.
Oliveira, Eliezer Fernando; Autreto, Pedro Alves da Silva; Galvao, Douglas Soares
On hardening silver nanocubes by high velocity impacts: a fully atomistic molecular dynamics investigation Journal Article
In: Journal of Materials Science, vol. 53, no. 10, pp. 7486–7492, 2018.
Abstract | Links | BibTeX | Tags: Fracture, impact, Molecular Dynamics, silver
@article{Oliveira2018,
title = {On hardening silver nanocubes by high velocity impacts: a fully atomistic molecular dynamics investigation},
author = {Oliveira, Eliezer Fernando and Autreto, Pedro Alves da Silva and Galvao, Douglas Soares},
url = {https://link.springer.com/article/10.1007/s10853-018-2104-z},
doi = {10.1007/s10853-018-2104-z},
year = {2018},
date = {2018-02-09},
journal = {Journal of Materials Science},
volume = {53},
number = {10},
pages = {7486–7492},
abstract = {Gradient nanograins (GNG) creation in metals has been a promising approach to obtain ultra-strong materials. Recently, R. Thevamaran et al. (Science 354:312 in 2016) proposed a single-step method based on high-velocity impacts of silver nanocubes (SNC) to produce almost perfect GNG. However, after certain time, these grains spontaneously coalesce, which compromises the induced hardening and other mechanical properties. To better understand these processes, a detailed investigation at the atomic scale of the deformation/hardening mechanisms are needed, which is one of the objectives of the present work. We carried out fully atomistic molecular dynamics (MD) simulations of silver nanocubes at high impact velocity values using realistic structural models. Our MD results suggest that besides the GNG mechanisms, the observed SNC hardening could be also the result of the existence of polycrystalline arrangements formed by HCP domains encapsulated by FCC ones in the smashed SNC. This can be a new way to design ultra-strong materials, even in the absence of GNG domains.},
keywords = {Fracture, impact, Molecular Dynamics, silver},
pubstate = {published},
tppubtype = {article}
}
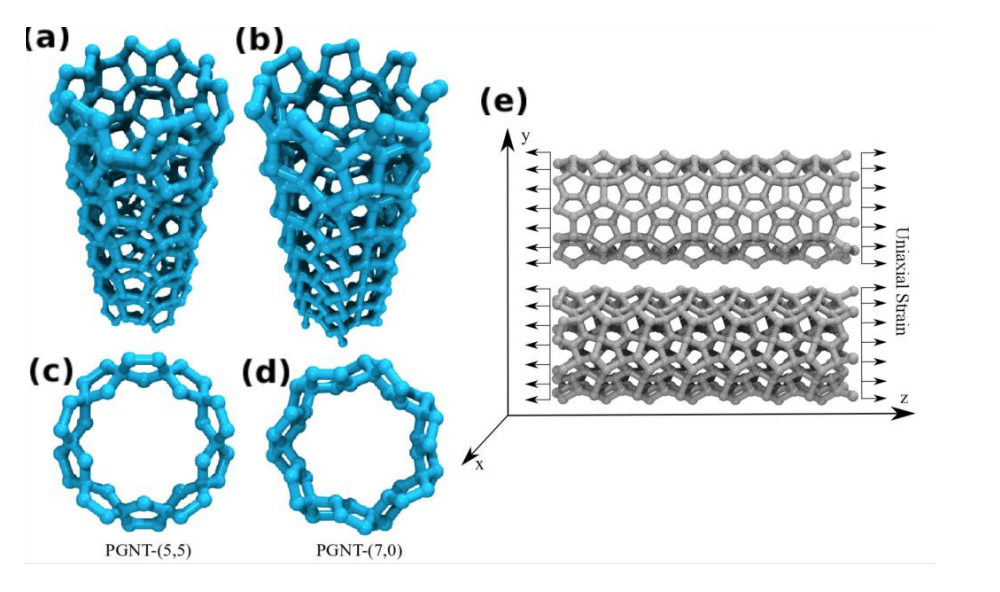
de Sousa, J. M.; Aguiar, A. L.; Girao, E. C.; Fonseca, Alexandre F.; Filho, A. G. Souza; Galvao, Douglas S.
Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 97-102, 2018.
Abstract | Links | BibTeX | Tags: Fracture, Molecular Dynamics, pentagraphene
@article{deSousa2018b,
title = {Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study},
author = {J. M. de Sousa and A. L. Aguiar and E. C. Girao and Alexandre F. Fonseca and A. G. Souza Filho and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-pentagraphenebased-nanotubes-a-molecular-dynamics-study/289AB70DADF20059BB8FCC9EF07B97AB},
doi = { https://doi.org/10.1557/adv.2018.160},
year = {2018},
date = {2018-02-06},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = {97-102},
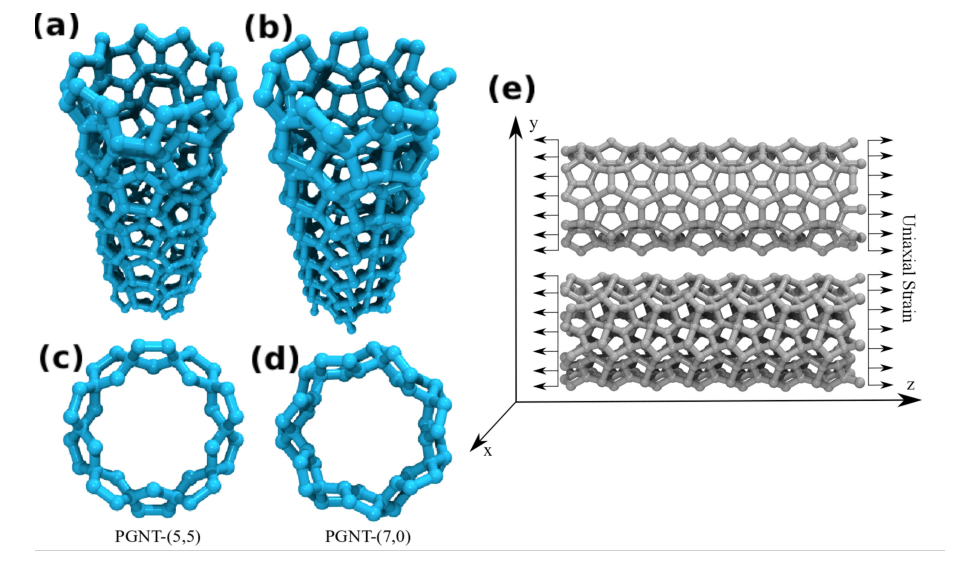
abstract = {The study of the mechanical properties of nanostructured systems has gained importance in theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of the strongest nanomaterials found in nature, with Young’s Modulus (EY) in the order 1.25 TPa. One interesting question is about the possibility of generating new nanostructures with 1D symmetry and with similar and/or superior CNT properties. In this work, we present a study on the dynamical, structural, mechanical properties, fracture patterns and EY values for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized states) in the same form that CNTs are formed from rolling up graphene membranes. We carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We have considered zigzag-like and armchair-like PGNTs of different diameters. Our results show that PGNTs present EY ∼ 800 GPa with distinct elastic behavior in relation to CNTs, mainly associated with mechanical failure, chirality dependent fracture patterns and extensive structural reconstructions.},
keywords = {Fracture, Molecular Dynamics, pentagraphene},
pubstate = {published},
tppubtype = {article}
}
Fonseca, Alexandre F.; Galvao, Douglas S.
Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation Journal Article
In: MRS Advances, vol. 3, no. 8-9, pp. 460-465, 2018.
Abstract | Links | BibTeX | Tags: Fracture, Graphene, Molecular Dynamics
@article{Fonseca2018,
title = {Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation},
author = {Alexandre F. Fonseca and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/selfdriven-graphene-tearing-and-peeling-a-fully-atomistic-molecular-dynamics-investigation/BFC76FC4479AA617E16FA6AC7AB4D487},
doi = {https://doi.org/10.1557/adv.2018.120},
year = {2018},
date = {2018-01-30},
journal = {MRS Advances},
volume = {3},
number = {8-9},
pages = {460-465},
abstract = {In spite of years of intense research, graphene continues to produce surprising results. Recently, it was experimentally observed that under certain conditions graphene can self-drive its tearing and peeling from substrates. This process can generate long, micrometer sized, folded nanoribbons without the action of any external forces. Also, during this cracking-like propagation process, the width of the graphene folded ribbon continuously decreases and the process only stops when the width reaches about few hundreds nanometers in size. It is believed that interplay between the strain energy of folded regions, breaking of carbon-carbon covalent bonds, and adhesion of graphene-graphene and graphene-substrate are the most fundamental features of this process, although the detailed mechanisms at atomic scale remain unclear. In order to gain further insights on these processes we carried out fully atomistic reactive molecular dynamics simulations using the AIREBO potential as available in the LAMMPS computational package. Although the reported tearing/peeling experimental observations were only to micrometer sized structures, our results showed that they could also occur at nanometer scale. Our preliminary results suggest that the graphene tearing/peeling process originates from thermal energy fluctuations that results in broken bonds, followed by strain release that creates a local elastic wave that can either reinforce the process, similar to a whip cracking propagation, or undermine it by producing carbon dangling bonds that evolve to the formation of bonds between the two layers of graphene. As the process continues in time and the folded graphene decreases in width, the carbon-carbon bonds at the ribbon edge and interlayer bonds get less stressed, thermal fluctuations become unable to break them and the process stops.},
keywords = {Fracture, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Azevedo, David L.; Bizao, Rafael A.; Galvao, Douglas S.
Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets Online
2018, (preprint arXiv:1801.05346).
Abstract | Links | BibTeX | Tags: Fracture, Molecular Dynamics, pentagraphene
@online{Azevedo2018b,
title = {Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets},
author = {David L. Azevedo and Rafael A. Bizao and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.05346},
year = {2018},
date = {2018-01-18},
abstract = {The superior mechanical properties and low density of carbon nanostructures make them promising ballistic protection materials, stimulating investigations on their high-strain-rate behavior. Recent experiments and simulations revealed graphene possesses exceptional energy absorption properties. In this work, we analyzed through fully atomistic molecular dynamics simulations the ballistic performance of a carbon-based material recently proposed named penta-graphene. Our results show that the fracture pattern is more spherical (no petals formation like observed for graphene). The estimated penetration energy for pentagraphene structures considered here was of 37.69 MJ/Kg, far superior to graphene (29.8 MJ/Kg) under same conditions. These preliminary results are suggestive that pentagraphene could be an excellent material for ballistic applications.},
note = {preprint arXiv:1801.05346},
keywords = {Fracture, Molecular Dynamics, pentagraphene},
pubstate = {published},
tppubtype = {online}
}
Fonseca, Alexandre F.; Galvao, Douglas S.
Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation Online
2018, (preprint arXiv:1801.05354).
Abstract | Links | BibTeX | Tags: Fracture, Graphene, Molecular Dynamics
@online{Fonseca2018b,
title = {Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation},
author = {Alexandre F. Fonseca and Douglas S. Galvao
},
url = {https://arxiv.org/abs/1801.05354},
year = {2018},
date = {2018-01-17},
abstract = {In spite of years of intense research, graphene continues to produce surprising results. Recently, it was experimentally observed that under certain conditions graphene can self-drive its tearing and peeling from substrates. This process can generate long, micrometer sized, folded nanoribbons without the action of any external forces. Also, during this cracking-like propagation process, the width of the graphene folded ribbon continuously decreases and the process only stops when the width reaches about few hundreds nanometers in size. It is believed that interplay between the strain energy of folded regions, breaking of carbon-carbon covalent bonds, and adhesion of graphene-graphene and graphene-substrate are the most fundamental features of this process, although the detailed mechanisms at atomic scale remain unclear. In order to gain further insights on these processes we carried out fully atomistic reactive molecular dynamics simulations using the AIREBO potential as available in the LAMMPS computational package. Although the reported tearing/peeling experimental observations were only to micrometer sized structures, our results showed that they could also occur at nanometer scale. Our preliminary results suggest that the graphene tearing/peeling process originates from thermal energy fluctuations that results in broken bonds, followed by strain release that creates a local elastic wave that can either reinforce the process, similar to a whip cracking propagation, or undermine it by producing carbon dangling bonds that evolve to the formation of bonds between the two layers of graphene. As the process continues in time and the folded graphene decreases in width, the carbon-carbon bonds at the ribbon edge and interlayer bonds get less stressed, thermal fluctuations become unable to break them and the process stops.},
note = {preprint arXiv:1801.05354},
keywords = {Fracture, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}
de Sousa, J. M.; Aguiar, A. L.; Girao, E. C.; Fonseca, Alexandre F.; Filho, A. G. Souza; Galvao, Douglas S.
Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 67-72, 2018.
Abstract | Links | BibTeX | Tags: Fracture, Molecular Dynamics, phagraphene
@article{deSousa2018c,
title = {Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation},
author = {J. M. de Sousa and A. L. Aguiar and E. C. Girao and Alexandre F. Fonseca and A. G. Souza Filho and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-phagraphene-membranes-a-fully-atomistic-molecular-dynamics-investigation/3ADC3F3B0052AB6632E8681404948E7B},
doi = {DOI: 10.1557/adv.2018. 54},
year = {2018},
date = {2018-01-15},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = {67-72},
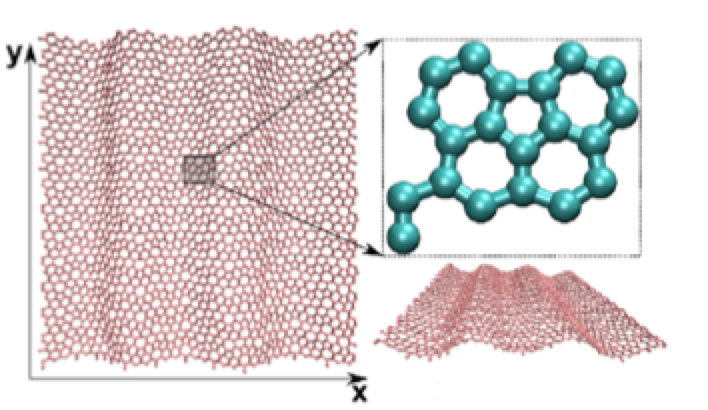
abstract = {Recently, a new 2D carbon allotrope structure, named phagraphene (PG), was proposed. PG has a densely array of penta-hexa-hepta-graphene carbon rings. PG was shown to present low and anisotropic thermal conductivity and it is believed that this anisotropy should be also reflected in its mechanical properties. Although PG mechanical properties have been investigated, a detailed and comprehensive study is still lacking. In the present work we have carried out fully atomistic reactive molecular dynamics simulations using the ReaxFF force field, to investigate the mechanical properties and fracture patterns of PG membranes. The Young's modulus values of the PG membranes were estimated from the stress-strain curves. Our results show that these curves present three distinct regimes: one regime where ripples dominate the structure and mechanical properties of the PG membranes; an elastic regime where the membranes exhibit fully planar configurations; and finally am inelastic regime where permanent deformations happened to the PG membrane up to the mechanical failure or fracture.},
keywords = {Fracture, Molecular Dynamics, phagraphene},
pubstate = {published},
tppubtype = {article}
}
de Sousa, J. M.; Aguiar, A. L.; Girao, E. C.; Fonseca, Alexandre F.; Filho, A. G. Sousa; Galvao, Douglas S.
Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study Online
2018, (preprint arXiv:1801.04269).
Abstract | Links | BibTeX | Tags: Fracture, Molecular Dynamics, Nanotubes, pentagraphene
@online{deSousa2018d,
title = {Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study},
author = {J. M. de Sousa and A. L. Aguiar and E. C. Girao and Alexandre F. Fonseca and A. G. Sousa Filho and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.04269},
year = {2018},
date = {2018-01-15},
abstract = {The study of the mechanical properties of nanostructured systems has gained importance in
theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of
the strongest nanomaterials found in nature, with Young's Modulus (YM) in the order 1.25
TPa. One interesting question is about the possibility of generating new nanostructures with
1D symmetry and with similar and/or superior CNT properties. In this work, we present a
study on the dynamical, structural, mechanical properties, fracture patterns and YM values
for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These
tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional
structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized
states) in the same form that CNTs are formed from rolling up graphene membranes. We
carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We
have considered zigzag-like and armchair-like PGNTs of different diameters. Our results
show that PGNTs present YM ~ 800 GPa with distinct elastic behavior in relation to CNTs,
mainly associated with mechanical failure, chirality dependent fracture patterns and extensive
structural reconstructions},
note = {preprint arXiv:1801.04269},
keywords = {Fracture, Molecular Dynamics, Nanotubes, pentagraphene},
pubstate = {published},
tppubtype = {online}
}
theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of
the strongest nanomaterials found in nature, with Young's Modulus (YM) in the order 1.25
TPa. One interesting question is about the possibility of generating new nanostructures with
1D symmetry and with similar and/or superior CNT properties. In this work, we present a
study on the dynamical, structural, mechanical properties, fracture patterns and YM values
for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These
tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional
structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized
states) in the same form that CNTs are formed from rolling up graphene membranes. We
carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We
have considered zigzag-like and armchair-like PGNTs of different diameters. Our results
show that PGNTs present YM ~ 800 GPa with distinct elastic behavior in relation to CNTs,
mainly associated with mechanical failure, chirality dependent fracture patterns and extensive
structural reconstructions

de Sousa, Jose M.; Aguiar, Acrisio L.; Girao, Eduardo C.; Fonseca, Alexandre F.; Antonio G. Sousa Filho,; Galvao, Douglas S.
Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation Online
2018, (preprint arXiv:1801.04292).
Abstract | Links | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, phagraphene
@online{deSousa2018e,
title = {Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Jose M. de Sousa and Acrisio L. Aguiar and Eduardo C. Girao and Alexandre F. Fonseca and Antonio G. Sousa Filho, and Douglas S. Galvao
},
url = {https://arxiv.org/abs/1801.04292},
year = {2018},
date = {2018-01-12},
abstract = {Recently, a new 2D carbon allotrope structure, named phagraphene (PG), was proposed. PG has a densely array of penta-hexa-hepta-graphene carbon rings. PG was shown to present low and anisotropic thermal conductivity and it is believed that this anisotropy should be also reflected in its mechanical properties. Although PG mechanical properties have been investigated, a detailed and comprehensive study is still lacking. In the present work we have carried out fully atomistic reactive molecular dynamics simulations using the ReaxFF force field, to investigate the mechanical properties and fracture patterns of PG membranes. The Young's modulus values of the PG membranes were estimated from the stress-strain curves. Our results show that these curves present three distinct regimes: one regime where ripples dominate the structure and mechanical properties of the PG membranes; an elastic regime where the membranes exhibit fully planar configurations; and finally a plastic regime where permanent deformations happened to the PG membrane up to the mechanical failure or fracture.},
note = {preprint arXiv:1801.04292},
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, phagraphene},
pubstate = {published},
tppubtype = {online}
}
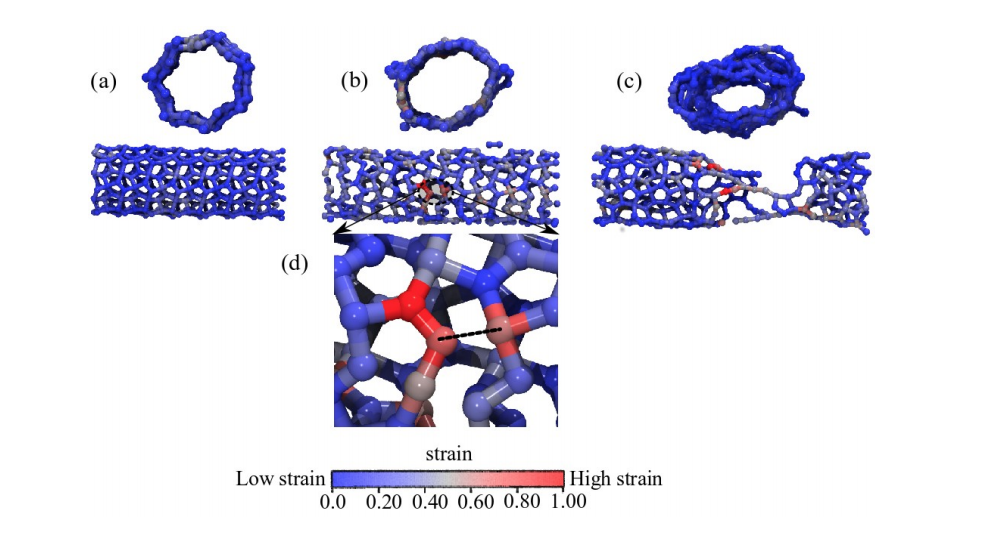
de Sousa, Jose M.; Aguiar, Acrisio L.; Girao, Eduardo C.; Fonseca, Alexandre F.; Antonio G. Souza Filho,; Galvao, Douglas S.
Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study Online
2018, (preprint arXiv:1801.04269).
Abstract | Links | BibTeX | Tags: Fracture, Molecular Dynamics, pentagraphene
@online{deSousa2018f,
title = {Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study},
author = {Jose M. de Sousa and Acrisio L. Aguiar and Eduardo C. Girao and Alexandre F. Fonseca and Antonio G. Souza Filho, and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.04269},
year = {2018},
date = {2018-01-12},
abstract = {The study of the mechanical properties of nanostructured systems has gained importance in theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of the strongest nanomaterials found in nature, with Young's Modulus (YM) in the order 1.25 TPa. One interesting question is about the possibility of generating new nanostructures with 1D symmetry and with similar and/or superior CNT properties. In this work, we present a study on the dynamical, structural, mechanical properties, fracture patterns and YM values for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized states) in the same form that CNTs are formed from rolling up graphene membranes. We carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We have considered zigzag-like and armchair-like PGNTs of different diameters. Our results show that PGNTs present YM ~ 800 GPa with distinct elastic behavior in relation to CNTs, mainly associated with mechanical failure, chirality dependent fracture patterns and extensive structural reconstructions.},
note = {preprint arXiv:1801.04269},
keywords = {Fracture, Molecular Dynamics, pentagraphene},
pubstate = {published},
tppubtype = {online}
}

Azevedo, David L.; Bizao, Rafael A.; Galvao, Douglas S.
Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets Journal Article
In: MRS Advances, vol. 3, no. 8-9, pp. 431-435, 2018.
Abstract | Links | BibTeX | Tags: Fracture, Molecular Dynamics, pentagraphene
@article{Azevedo2018,
title = {Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets},
author = {David L. Azevedo and Rafael A. Bizao and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/molecular-dynamics-simulations-of-ballistic-penetration-of-pentagraphene-sheets/8759C0815840EDE83896EF4A17278228},
doi = {https://doi.org/10.1557/adv.2018.61},
year = {2018},
date = {2018-01-06},
journal = {MRS Advances},
volume = {3},
number = {8-9},
pages = {431-435},
abstract = {The search for new materials with low density and superior mechanical properties is a very intense and stimulating investigation area. These new materials could provide potential application for ballistic protection. Recent experiments and simulations revealed graphene possesses exceptional energy absorption properties. In this work, we analysed through fully atomistic molecular dynamics simulations the ballistic performance of a carbon-based material recently proposed named penta-graphene. Our results show that the fracture pattern is more spherical (no petals formation like observed for graphene). The estimated penetration energy for single-layer penta-graphene structures obtained here was d_1penta∼37.7 MJ/kg, and is comparable with recently results obtained for graphene: d_(1graphene)∼29.0 MJ/kg and d_(1graphene)∼40.8 MJ/kg under similar conditions. These preliminary results are suggestive that penta-graphene could be an excellent material for ballistic applications.},
keywords = {Fracture, Molecular Dynamics, pentagraphene},
pubstate = {published},
tppubtype = {article}
}
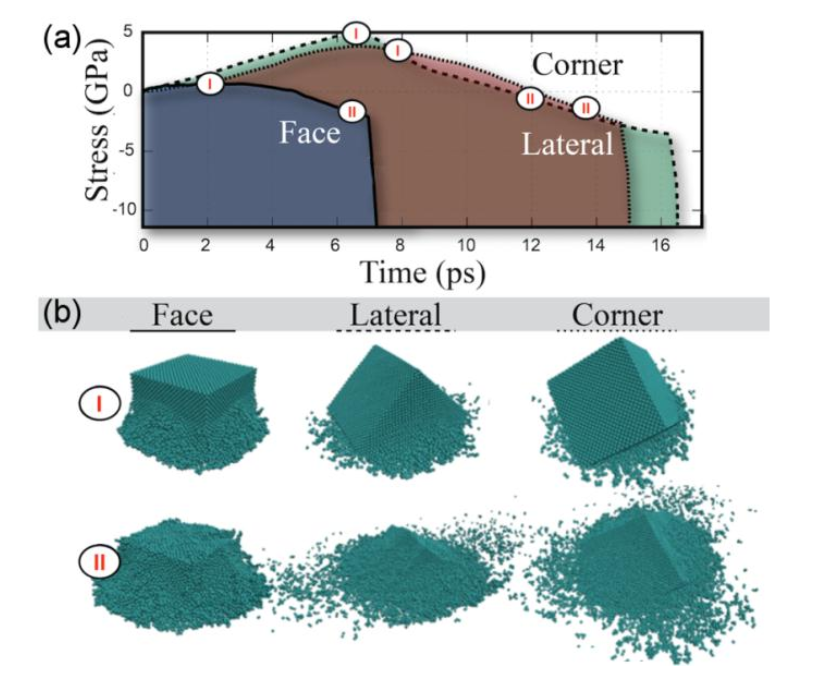
Oliveira, Eliezer Fernando; da Silva Autreto, Pedro Alves; Galvao, Douglas Soares
Silver Hardening via Hypersonic Impacts Journal Article
In: MRS Advances, vol. 3, no. 8-9, pp. 489-494, 2018.
Abstract | Links | BibTeX | Tags: Fracture, impact, Molecular Dynamics, silver
@article{Oliveira2018b,
title = {Silver Hardening via Hypersonic Impacts},
author = {Eliezer Fernando Oliveira and Pedro Alves da Silva Autreto and Douglas Soares Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/silver-hardening-via-hypersonic-impacts/6A35FAB117B4FD244BBD11A64CD25160},
doi = {DOI: 10.1557/adv.2018. 173},
year = {2018},
date = {2018-01-01},
journal = {MRS Advances},
volume = {3},
number = {8-9},
pages = {489-494},
abstract = {The search for new ultra strong materials has been a very active research area. With relation to metals, a successful way to improve their strength is by the creation of a gradient of nanograins (GNG) inside the material. Recently, R. Thevamaran et al. [Science v354, 312- 316 (2016)] propose a single step method based on high velocity impact of silver nanocubes to produce high-quality GNG. This method consists of producing high impact collisions of silver cubes at hypersonic velocity (~400 m/s) against a rigid wall. Although they observed an improvement in the mechanical properties of the silver after the impact, the GNG creation and the strengthening mechanism at nanoscale remain unclear. In order to gain further insights about these mechanisms, we carried out fully atomistic molecular dynamics simulations (MD) to investigate the atomic conformations/rearrangements during and after high impact collisions of silver nanocubes at ultrasonic velocity. Our results indicate the co- existence of polycrystalline arrangements after the impact formed by core HCP domains surrounded by FCC ones, which could also contribute to explain the structural hardening.},
keywords = {Fracture, impact, Molecular Dynamics, silver},
pubstate = {published},
tppubtype = {article}
}
2017
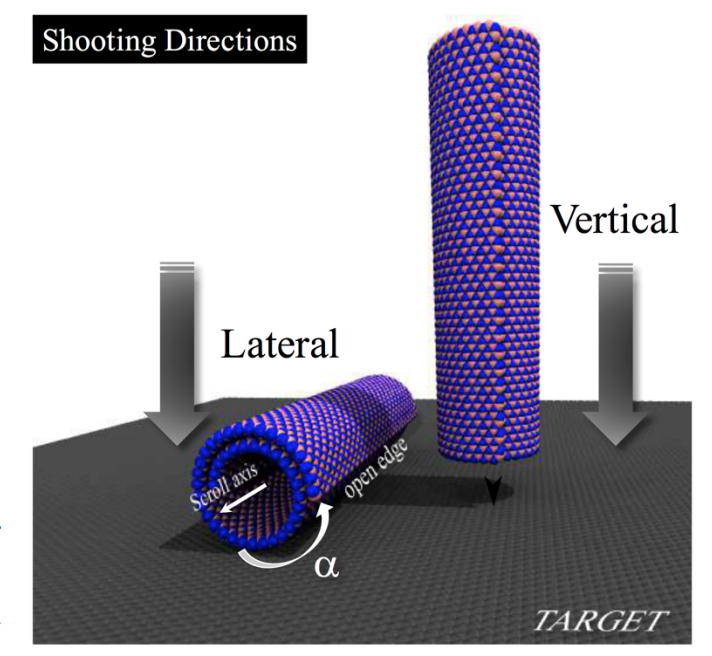
Leonardo D Machado Cristiano F Woellner, Pedro AS Autreto; Galvao, Douglas S
Structural Transformations of Carbon and Boron Nitride Nanoscrolls at High Impact Collisions Online
2017, (preprint ArXiv:1711.00378).
Abstract | Links | BibTeX | Tags: Fracture, impacts, Molecular Dynamics, Scrolls
@online{Woellner2017,
title = {Structural Transformations of Carbon and Boron Nitride Nanoscrolls at High Impact Collisions},
author = {Cristiano F Woellner, Leonardo D Machado, Pedro AS Autreto, Jose M de Sousa, and Douglas S Galvao},
url = {https://arxiv.org/pdf/1711.00378.pdf},
year = {2017},
date = {2017-11-01},
abstract = {The behavior of nanostructures under high strain-rate conditions has been object of theoretical and experimental investigations in recent years. For instance, it has been shown that carbon and boron nitride nanotubes can be unzipped into nanoribbons at high velocity impacts. However, the response of many nanostructures to high strain-rate conditions is still not completely understood. In this work we have investigated through fully atomistic reactive (ReaxFF) molecular dynamics (MD) simulations the mechanical behavior of carbon (CNS) and boron nitride nanoscrolls (BNS) colliding against solid targets at high velocities,. CNS (BNS) nanoscrolls are graphene (boron nitride) membranes rolled up into papyrus-like
structures. Their open-ended topology leads to unique properties not found in close-ended analogues, such as nanotubes.Our results show that the collision products are mainly determined by impact velocities and by two impact angles, which
define the position of the scroll (i) axis and (ii) open edge relative to the target. Our MD results showed that for appropriate velocities and orientations large-scale deformations and nanoscroll fracture can occur. We also observed unscrolling (scrolls going back to quasi-planar membranes), scroll unzipping into nanoribbons, and significant
reconstruction due to breaking and/or formation of new chemical bonds. For particular edge orientations and velocities, conversion from open to close-ended topology is also possible, due to the fusion of nanoscroll walls.},
note = {preprint ArXiv:1711.00378},
keywords = {Fracture, impacts, Molecular Dynamics, Scrolls},
pubstate = {published},
tppubtype = {online}
}
structures. Their open-ended topology leads to unique properties not found in close-ended analogues, such as nanotubes.Our results show that the collision products are mainly determined by impact velocities and by two impact angles, which
define the position of the scroll (i) axis and (ii) open edge relative to the target. Our MD results showed that for appropriate velocities and orientations large-scale deformations and nanoscroll fracture can occur. We also observed unscrolling (scrolls going back to quasi-planar membranes), scroll unzipping into nanoribbons, and significant
reconstruction due to breaking and/or formation of new chemical bonds. For particular edge orientations and velocities, conversion from open to close-ended topology is also possible, due to the fusion of nanoscroll walls.

Bizao, Rafael A; Machado, Leonardo D; de Sousa, Jose M; Pugno, Nicola M; Galvao, Douglas S
Scale Effects on the Ballistic Penetration of Graphene Sheets Online
2017, (preprint arXiv:1701.07367).
Abstract | Links | BibTeX | Tags: ballistic impacts, Fracture, Graphene, Molecular Dynamics
@online{Bizao2017c,
title = {Scale Effects on the Ballistic Penetration of Graphene Sheets},
author = {Bizao, Rafael A and Machado, Leonardo D and de Sousa, Jose M and Pugno, Nicola M and Galvao, Douglas S},
url = {https://arxiv.org/pdf/1701.07367.pdf},
year = {2017},
date = {2017-01-25},
abstract = {Carbon nanostructures are promising ballistic protection materials,
due to their low density and excellent mechanical properties. Recent
experimental and computational investigations on the behavior
of graphene under impact conditions revealed exceptional energy absorption
properties as well. However, the reported numerical and experimental
values differ by an order of magnitude. In this work, we
combined numerical and analytical modeling to address this issue. In
the numerical part, we employed reactive molecular dynamics to carry
out ballistic tests on single and double-layered graphene sheets. We
used velocity values within the range tested in experiments. Our numerical
and the experimental results were used to determine parameters
for a scaling law, which is in good agreement with all experimental
and simulation results. We find that the specific penetration energy
decreases as the number of layers (N) increases, from ∼ 25 MJ/kg for
N = 1 to ∼ 0.26 MJ/kg as N → ∞. These scale effects explain the
apparent discrepancy between simulations and experiments.},
note = {preprint arXiv:1701.07367},
keywords = {ballistic impacts, Fracture, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}
due to their low density and excellent mechanical properties. Recent
experimental and computational investigations on the behavior
of graphene under impact conditions revealed exceptional energy absorption
properties as well. However, the reported numerical and experimental
values differ by an order of magnitude. In this work, we
combined numerical and analytical modeling to address this issue. In
the numerical part, we employed reactive molecular dynamics to carry
out ballistic tests on single and double-layered graphene sheets. We
used velocity values within the range tested in experiments. Our numerical
and the experimental results were used to determine parameters
for a scaling law, which is in good agreement with all experimental
and simulation results. We find that the specific penetration energy
decreases as the number of layers (N) increases, from ∼ 25 MJ/kg for
N = 1 to ∼ 0.26 MJ/kg as N → ∞. These scale effects explain the
apparent discrepancy between simulations and experiments.
Oliveira, Eliezer Fernando; Pedro Alves da Silva Autreto,; Galvao, Douglas Soares
Silver Hardening via Hypersonic Impacts Online
2017, (preprint arXiv:1801.04780).
Abstract | Links | BibTeX | Tags: Fracture, impact, Molecular Dynamics, silver
@online{Oliveira2017,
title = {Silver Hardening via Hypersonic Impacts},
author = {Eliezer Fernando Oliveira and Pedro Alves da Silva Autreto, and Douglas Soares Galvao},
url = {https://arxiv.org/abs/1801.04780},
year = {2017},
date = {2017-01-15},
abstract = {The search for new ultra strong materials has been a very active research area. With relation
to metals, a successful way to improve their strength is by the creation of a gradient of
nanograins (GNG) inside the material. Recently, R. Thevamaran et al. [Science v354, 312-
316 (2016)] propose a single step method based on high velocity impact of silver nanocubes
to produce high-quality GNG. This method consists of producing high impact collisions of
silver cubes at hypersonic velocity (~400 m/s) against a rigid wall. Although they observed an
improvement in the mechanical properties of the silver after the impact, the GNG creation
and the strengthening mechanism at nanoscale remain unclear. In order to gain further
insights about these mechanisms, we carried out fully atomistic molecular dynamics
simulations (MD) to investigate the atomic conformations/rearrangements during and after
high impact collisions of silver nanocubes at ultrasonic velocity. Our results indicate the coexistence
of polycrystalline arrangements after the impact formed by core HCP domains
surrounded by FCC ones, which could also contribute to explain the structural hardening.},
note = {preprint arXiv:1801.04780},
keywords = {Fracture, impact, Molecular Dynamics, silver},
pubstate = {published},
tppubtype = {online}
}
to metals, a successful way to improve their strength is by the creation of a gradient of
nanograins (GNG) inside the material. Recently, R. Thevamaran et al. [Science v354, 312-
316 (2016)] propose a single step method based on high velocity impact of silver nanocubes
to produce high-quality GNG. This method consists of producing high impact collisions of
silver cubes at hypersonic velocity (~400 m/s) against a rigid wall. Although they observed an
improvement in the mechanical properties of the silver after the impact, the GNG creation
and the strengthening mechanism at nanoscale remain unclear. In order to gain further
insights about these mechanisms, we carried out fully atomistic molecular dynamics
simulations (MD) to investigate the atomic conformations/rearrangements during and after
high impact collisions of silver nanocubes at ultrasonic velocity. Our results indicate the coexistence
of polycrystalline arrangements after the impact formed by core HCP domains
surrounded by FCC ones, which could also contribute to explain the structural hardening.
2016
Chandra Sekhar Tiwary Mohamad A Kabbani, Anirban Som
A generic approach for mechano-chemical reactions between carbon nanotubes of different functionalities Journal Article
In: Carbon, vol. 104, pp. 196-202, 2016.
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, DFT, Fracture, Mechano-chemistry, Molecular Dynamics
@article{Kabbani2016,
title = {A generic approach for mechano-chemical reactions between carbon nanotubes of different functionalities},
author = {Mohamad A Kabbani, Chandra Sekhar Tiwary, Anirban Som, KR Krishnadas, Pedro AS Autreto, Sehmus Ozden, Kunttal Keyshar, Ken Hackenberg, Alin Christian Chipara, Douglas S Galvao, Robert Vajtai, Ahmad T Kabbani, Thalappil Pradeep, Pulickel M Ajayan},
url = {www.sciencedirect.com/science/article/pii/S000862231630183X},
doi = {10.1016/j.carbon.2016.02.094},
year = {2016},
date = {2016-08-31},
journal = {Carbon},
volume = {104},
pages = {196-202},
abstract = {Abstract Here, we report similar reactions between nanotubes carrying functionalities,
namely carbon nanotubes (CNTs) with the acyl chloride/hydroxyl and amine/carboxylic
functionalities directly attached to their surfaces, resulting in the formation ofchemically
modified graphene products. The reaction is spontaneous and is facilitated by simple
grinding of the reactants. The new solid-state reactions have been confirmed using different
spectroscopic and electron microscopy techniques.},
keywords = {Carbon Nanotubes, DFT, Fracture, Mechano-chemistry, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
namely carbon nanotubes (CNTs) with the acyl chloride/hydroxyl and amine/carboxylic
functionalities directly attached to their surfaces, resulting in the formation ofchemically
modified graphene products. The reaction is spontaneous and is facilitated by simple
grinding of the reactants. The new solid-state reactions have been confirmed using different
spectroscopic and electron microscopy techniques.
Sehmus Ozden Leonardo D Machado, ChandraSekhar Tiwary
The structural and dynamical aspects of boron nitride nanotubes under high velocity impacts Journal Article
In: Physical Chemistry Chemical Physics, vol. 18, pp. 14776-14781, 2016.
Abstract | Links | BibTeX | Tags: Ballistic Impact, Boron Nitride tubes, CNT, Fracture, Molecular Dynamics
@article{Machado2016,
title = {The structural and dynamical aspects of boron nitride nanotubes under high velocity impacts},
author = {Leonardo D Machado, Sehmus Ozden, ChandraSekhar Tiwary, Pedro AS Autreto, Robert Vajtai, Enrique V Barrera, Douglas S Galvao, Pulickel M Ajayan},
url = {xlink.rsc.org/?DOI=c6cp01949h},
doi = {10.1039/C6CP01949H},
year = {2016},
date = {2016-05-01},
journal = {Physical Chemistry Chemical Physics},
volume = {18},
pages = {14776-14781},
abstract = {This communication report is a study on the structural and dynamical aspects of boron nitride nanotubes (BNNTs) shot at high velocities (∼5 km s−1) against solid targets. The experimental results show unzipping of BNNTs and the formation of hBN nanoribbons. Fully atomistic reactive molecular dynamics simulations were also carried out to gain insights into the BNNT fracture patterns and deformation mechanisms. Our results show that longitudinal and axial tube fractures occur, but the formation of BN nanoribbons from fractured tubes was only observed for some impact angles. Although some structural and dynamical features of the impacts are similar to the ones reported for CNTs, because BNNTs are more brittle than CNTs this results in a larger number of fractured tubes but with fewer formed nanoribbons.},
keywords = {Ballistic Impact, Boron Nitride tubes, CNT, Fracture, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
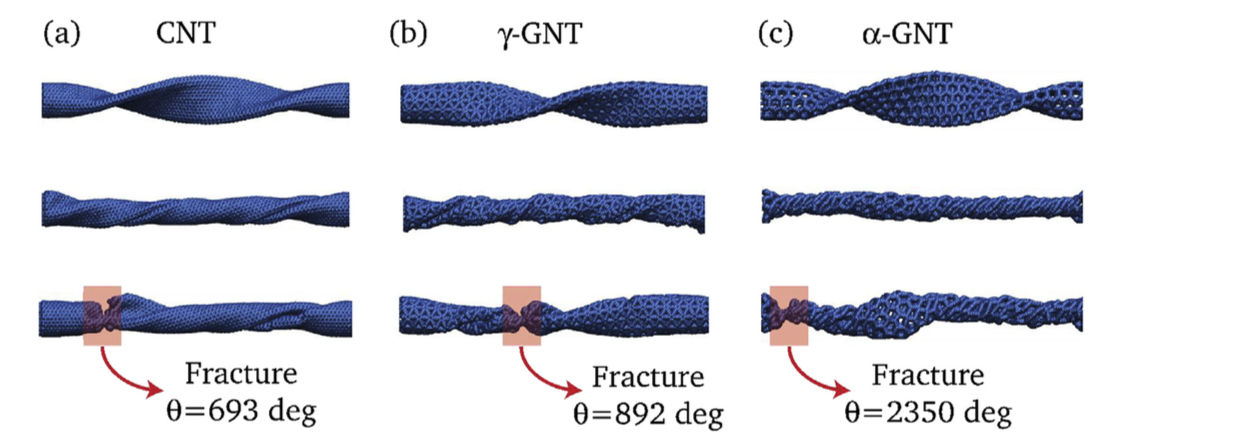
G. Brunetto J.M. de Sousa, V. R. Coluci
Torsional “superplasticity” of graphyne nanotubes Journal Article
In: Carbon, vol. 96, pp. 14-19, 2016.
Abstract | Links | BibTeX | Tags: Fracture, Graphynes, Mechanical Properties, Nanotubes
@article{deSousa2016,
title = {Torsional “superplasticity” of graphyne nanotubes},
author = {J.M. de Sousa, G. Brunetto, V.R. Coluci, D.S. Galvao },
url = {http://www.sciencedirect.com/science/article/pii/S000862231530258X},
doi = { http://dx.doi.org/10.1016/j.carbon.2015.09.039},
year = {2016},
date = {2016-01-01},
journal = {Carbon},
volume = {96},
pages = {14-19},
abstract = {Graphyne is a planar two-dimensional carbon allotrope formed by atoms in sp, sp2, and sp3 hybridized states. Topologically graphyne nanotubes (GNTs) can be considered as cylindrically rolled up graphyne sheets, similarly as carbon nanotubes (CNTs) can be considered rolled up graphene sheets. Due to the presence of single, double, and triple bonds, GNTs exhibit porous sidewalls that can be exploited in many diverse applications. In this work, we investigated the mechanical behavior of GNTs under torsional strains through reactive molecular dynamics simulations. Our results show that GNTs are more flexible than CNTs and exhibit “superplasticit”, with fracture angles that are up to 35 times higher than the ones reported to CNTs. This GNT “superplastic” behavior can be explained in terms of irreversible recon- struction processes (mainly associated with the triple bonds) that occur during torsional strains.},
keywords = {Fracture, Graphynes, Mechanical Properties, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
2014
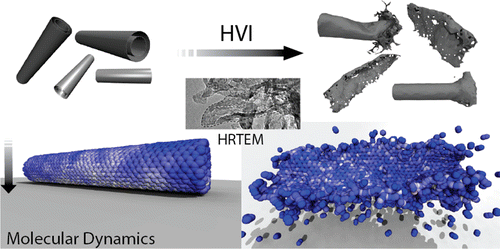
Ozden, Sehmus; Autreto, Pedro AS; Tiwary, Chandra Sekhar; Khatiwada, Suman; Machado, Leonardo; Galvao, Douglas S; Vajtai, Robert; Barrera, Enrique V; M. Ajayan, Pulickel
Unzipping Carbon Nanotubes at High Impact Journal Article
In: Nano letters, vol. 14, no. 7, pp. 4131–4137, 2014.
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, Fracture, Unzipping
@article{ozden2014unzipping,
title = {Unzipping Carbon Nanotubes at High Impact},
author = {Ozden, Sehmus and Autreto, Pedro AS and Tiwary, Chandra Sekhar and Khatiwada, Suman and Machado, Leonardo and Galvao, Douglas S and Vajtai, Robert and Barrera, Enrique V and M. Ajayan, Pulickel},
url = {http://pubs.acs.org/doi/abs/10.1021/nl501753n},
year = {2014},
date = {2014-01-01},
journal = {Nano letters},
volume = {14},
number = {7},
pages = {4131--4137},
publisher = {American Chemical Society},
abstract = {The way nanostructures behave and mechanically respond to high impact collision is a topic of intrigue. For anisotropic nanostructures, such as carbon nanotubes, this response will be complicated based on the impact geometry. Here we report the result of hypervelocity impact of nanotubes against solid targets and show that impact produces a large number of defects in the nanotubes, as well as rapid atom evaporation, leading to their unzipping along the nanotube axis. Fully atomistic reactive molecular dynamics simulations are used to gain further insights of the pathways and deformation and fracture mechanisms of nanotubes under high energy mechanical impact. Carbon nanotubes have been unzipped into graphene nanoribbons before using chemical treatments but here the instability of nanotubes against defect formation, fracture, and unzipping is revealed purely through mechanical impact.},
keywords = {Carbon Nanotubes, Fracture, Unzipping},
pubstate = {published},
tppubtype = {article}
}
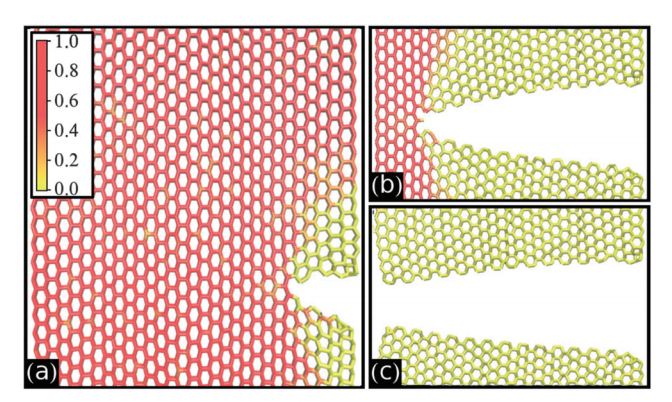
Botari, T; Perim, E; Autreto, PAS; van Duin, ACT; Paupitz, R; Galvao, DS
Mechanical properties and fracture dynamics of silicene membranes Journal Article
In: PHYSICAL CHEMISTRY CHEMICAL PHYSICS, vol. 16, no. 36, pp. 19417–19423, 2014.
Abstract | Links | BibTeX | Tags: Fracture, Germanene, Graphene, Mechanical Properties, Silicene
@article{botari2014mechanical,
title = {Mechanical properties and fracture dynamics of silicene membranes},
author = {Botari, T and Perim, E and Autreto, PAS and van Duin, ACT and Paupitz, R and Galvao, DS},
url = {http://pubs.rsc.org/en/content/articlehtml/2014/cp/c4cp02902j},
year = {2014},
date = {2014-01-01},
journal = {PHYSICAL CHEMISTRY CHEMICAL PHYSICS},
volume = {16},
number = {36},
pages = {19417--19423},
publisher = {ROYAL SOC CHEMISTRY},
abstract = {As graphene has become one of the most important materials, there is renewed interest in other similar structures. One example is silicene, the silicon analogue of graphene. It shares some of the remarkable graphene properties, such as the Dirac cone, but presents some distinct ones, such as a pronounced structural buckling. We have investigated, through density functional based tight-binding (DFTB), as well as reactive molecular dynamics (using ReaxFF), the mechanical properties of suspended single-layer silicene. We calculated the elastic constants, analyzed the fracture patterns and edge reconstructions. We also addressed the stress distributions, unbuckling mechanisms and the fracture dependence on the temperature. We analysed the differences due to distinct edge morphologies, namely zigzag and armchair.},
keywords = {Fracture, Germanene, Graphene, Mechanical Properties, Silicene},
pubstate = {published},
tppubtype = {article}
}

Vinod, Soumya; Tiwary, Chandra Sekhar; da Silva Autreto, Pedro Alves; Taha-Tijerina, Jaime; Ozden, Sehmus; Chipara, Alin Cristian; Vajtai, Robert; Galvao, Douglas S; Narayanan, Tharangattu N; Ajayan, Pulickel M
Low-density three-dimensional foam using self-reinforced hybrid two-dimensional atomic layers Journal Article
In: Nature Communications, vol. 5, 2014.
Links | BibTeX | Tags: foams, Fracture, Graphene, Mechanical Properties, top20
@article{vinod2014low,
title = {Low-density three-dimensional foam using self-reinforced hybrid two-dimensional atomic layers},
author = {Vinod, Soumya and Tiwary, Chandra Sekhar and da Silva Autreto, Pedro Alves and Taha-Tijerina, Jaime and Ozden, Sehmus and Chipara, Alin Cristian and Vajtai, Robert and Galvao, Douglas S and Narayanan, Tharangattu N and Ajayan, Pulickel M},
url = {http://www.nature.com/ncomms/2014/140729/ncomms5541/full/ncomms5541.html},
year = {2014},
date = {2014-01-01},
journal = {Nature Communications},
volume = {5},
publisher = {Nature Publishing Group},
keywords = {foams, Fracture, Graphene, Mechanical Properties, top20},
pubstate = {published},
tppubtype = {article}
}
2013

Perim, Eric; Santos, Ricardo Paupitz; Autreto, Pedro Alves da Silva; Galvao, Douglas S
Fracture Patterns of Boron Nitride Nanotubes Proceedings
Cambridge University Press, vol. 1526, 2013.
Abstract | Links | BibTeX | Tags: Boron Nitride, Fracture, Mechanical Properties, Unzipping
@proceedings{perim2013fracture,
title = {Fracture Patterns of Boron Nitride Nanotubes},
author = {Perim, Eric and Santos, Ricardo Paupitz and Autreto, Pedro Alves da Silva and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8883390&fileId=S1946427413004946},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1526},
pages = {mrsf12--1526},
publisher = {Cambridge University Press},
abstract = {During the last years carbon-based nanostructures (such as, fullerenes, carbon nanotubes and graphene) have been object of intense investigations. The great interest in these nanostructures can be attributed to their remarkable electrical and mechanical properties. Their inorganic equivalent structures do exist and are based on boron nitride (BN) motifs. BN fullerenes, nanotubes and single layers have been already synthesized. Recently, the fracture patterns of single layer graphene and multi-walled carbon nanotubes under stress have been studied by theoretical and experimental methods. In this work we investigated the fracturing process of defective carbon and boron nitride nanotubes under similar stress conditions. We have carried out fully atomistic molecular reactive molecular dynamics simulations using the ReaxFF force field. The similarities and differences between carbon and boron nitride fracture patterns are addressed.},
keywords = {Boron Nitride, Fracture, Mechanical Properties, Unzipping},
pubstate = {published},
tppubtype = {proceedings}
}
2012

Dos Santos, RPB; Perim, E; Autreto, PAS; Brunetto, Gustavo; Galvao, DS
On the unzipping of multiwalled carbon nanotubes Journal Article
In: Nanotechnology, vol. 23, no. 46, pp. 465702, 2012.
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, Fracture, Molecular Dynamics, Unzipping
@article{dos2012unzippingb,
title = {On the unzipping of multiwalled carbon nanotubes},
author = {Dos Santos, RPB and Perim, E and Autreto, PAS and Brunetto, Gustavo and Galvao, DS},
url = {http://iopscience.iop.org/0957-4484/23/46/465702},
year = {2012},
date = {2012-01-01},
journal = {Nanotechnology},
volume = {23},
number = {46},
pages = {465702},
publisher = {IOP Publishing},
abstract = {Graphene nanoribbons (GNRs) are very interesting structures which can retain graphene's high carrier mobility while presenting a finite bandgap. These properties make GNRs very valuable materials for the building of nanodevices. Unzipping carbon nanotubes (CNTs) is considered one of the most promising approaches for GNR controlled and large-scale production, although some of the details of the CNT unzipping processes are not completely known. In this work we have investigated CNT unzipping processes through fully atomistic molecular dynamics simulations using reactive force fields (ReaxFF). Multiwalled CNTs of different dimensions and chiralities under induced mechanical stretching were considered. Our results show that fracture patterns and stress profiles are highly CNT chirality dependent. Our results also show that the 'crests' (partially unzipped CNT regions presenting high curvature), originating from defective CNT areas, can act as a guide for the unzipping processes, which can explain the almost perfectly linear cuts frequently observed in unzipped CNTs.
},
keywords = {Carbon Nanotubes, Fracture, Molecular Dynamics, Unzipping},
pubstate = {published},
tppubtype = {article}
}
2007
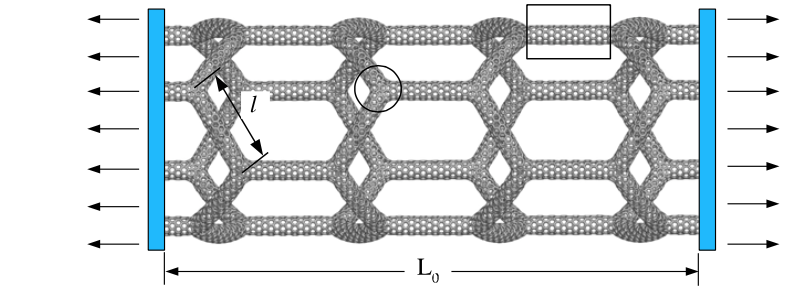
Coluci, Vitor R; Pugno, Nicola M; Dantas, Socrates O; Galvao, Douglas S; Jorio, Ado
Atomistic simulations of the mechanical properties of'super'carbon nanotubes Journal Article
In: Nanotechnology, vol. 18, no. 33, pp. 335702, 2007.
Abstract | Links | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, Super Carbons
@article{coluci2007atomistic,
title = {Atomistic simulations of the mechanical properties of'super'carbon nanotubes},
author = {Coluci, Vitor R and Pugno, Nicola M and Dantas, Socrates O and Galvao, Douglas S and Jorio, Ado},
url = {http://iopscience.iop.org/0957-4484/18/33/335702
},
year = {2007},
date = {2007-01-01},
journal = {Nanotechnology},
volume = {18},
number = {33},
pages = {335702},
publisher = {IOP Publishing},
abstract = {The mechanical properties of the so-called 'super' carbon nanotubes (STs) are investigated using classical molecular dynamics simulations. The STs are built from single-walled carbon nanotubes (SWCNTs) connected by Y-like junctions forming an ordered carbon nanotube network that is then rolled into a seamless cylinder. We observed that the ST behaviour under tensile tests is similar to the one presented by fishing nets. This interesting behaviour provides a way to vary the accessible channels to the inner parts of STs by applying an external mechanical load. The Young's modulus is dependent on the ST chirality and it inversely varies with the ST radius. Smaller reduction of breaking strain values due to temperature increase is predicted for zigzag STs compared to SWCNTs. The results show that, for STs with radius ~5 nm, the junctions between the constituent SWCNTs play an important role in the fracture process. The Young's modulus and tensile strength were estimated for hierarchical higher-order STs using scaling laws related to the ST fractal dimension. The obtained mechanical properties suggest that STs may be used in the development of new porous, flexible, and high-strength materials.},
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, Super Carbons},
pubstate = {published},
tppubtype = {article}
}
Coluci, VR; Dantas, SO; Jorio, A; Galvao, DS
Electronic and Mechanical Properties of Super Carbon Nanotube Networks Proceedings
Warrendale, Pa.; Materials Research Society; 1999, vol. 963, 2007.
Abstract | Links | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, Super Carbons
@proceedings{coluci2007electronic,
title = {Electronic and Mechanical Properties of Super Carbon Nanotube Networks},
author = {Coluci, VR and Dantas, SO and Jorio, A and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8026810&fulltextType=RA&fileId=S1946427400054014},
year = {2007},
date = {2007-01-01},
journal = {MATERIALS RESEARCH SOCIETY SYMPOSIUM PROCEEDINGS},
volume = {963},
pages = {1},
publisher = {Warrendale, Pa.; Materials Research Society; 1999},
abstract = {Eletronic and mechanical properties of ordered carbon nanotube networks are studied using molecular dynamics simulations and tight-binding calculations. These networks are formed by single walled carbon nanotubes (SWNT) regularly connected by junctions. The use of different types of junctions (“Y”-, “X”-like junctions, for example) allows the construction of networks with different symmetries. These networks can be very flexible and the elastic deformation was associated with two main deformation mechanisms (bending and stretching ) of the constituents SWNTs. Rolling up the networks, “super” carbon nanotubes can be constructed. These super-tubes share some of the main electronic features of the SWNT which form them but important changes are predicted (e.g. reduction of bandgap value). Simulations of their deformations under tensile stress have revealed that the super-tubes are softer than the corresponding SWNT and that their rupture occur in higher strain values.},
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, Super Carbons},
pubstate = {published},
tppubtype = {proceedings}
}
Coluci, VR; Dantas, SO; Jorio, A; Galvao, DS
Mechanical properties of carbon nanotube networks by molecular mechanics and impact molecular dynamics calculations Journal Article
In: Physical Review B, vol. 75, no. 7, pp. 075417, 2007.
Abstract | Links | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, Super Carbons
@article{coluci2007mechanical,
title = {Mechanical properties of carbon nanotube networks by molecular mechanics and impact molecular dynamics calculations},
author = {Coluci, VR and Dantas, SO and Jorio, A and Galvao, DS},
url = {http://journals.aps.org/prb/abstract/10.1103/PhysRevB.75.075417},
year = {2007},
date = {2007-01-01},
journal = {Physical Review B},
volume = {75},
number = {7},
pages = {075417},
publisher = {APS},
abstract = {We report a theoretical investigation of the mechanical properties of idealized networks formed by single-walled carbon nanotubes showing crossbar and hexagonal architectures. The study was performed by using molecular mechanics calculations and impact dynamics simulations based on bond-order empirical potential. The studied networks were predicted to have elasticity modulus of ∼10–100GPa and bulk modulus of ∼10GPa. The results show a transition from high to moderate flexibility during the deformation stages. This behavior was associated with the existence of two deformation mechanisms presented by the network related to the nanotube stretching and junction bending processes.},
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, Super Carbons},
pubstate = {published},
tppubtype = {article}
}
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ

