
Borges, Daiane Damasceno; Woellner, Cristiano F; Autreto, Pedro AS; Galvao, Douglas S
2017, (preprint arXiv:1702.00250).
@online{Borges2017b,
title = {Insights on the mechanism of water-alcohol separation in multilayer graphene oxide membranes: entropic versus enthalpic factors},
author = {Borges, Daiane Damasceno and Woellner, Cristiano F and Autreto, Pedro AS and Galvao, Douglas S},
url = {https://arxiv.org/abs/1706.06213},
year = {2017},
date = {2017-06-19},
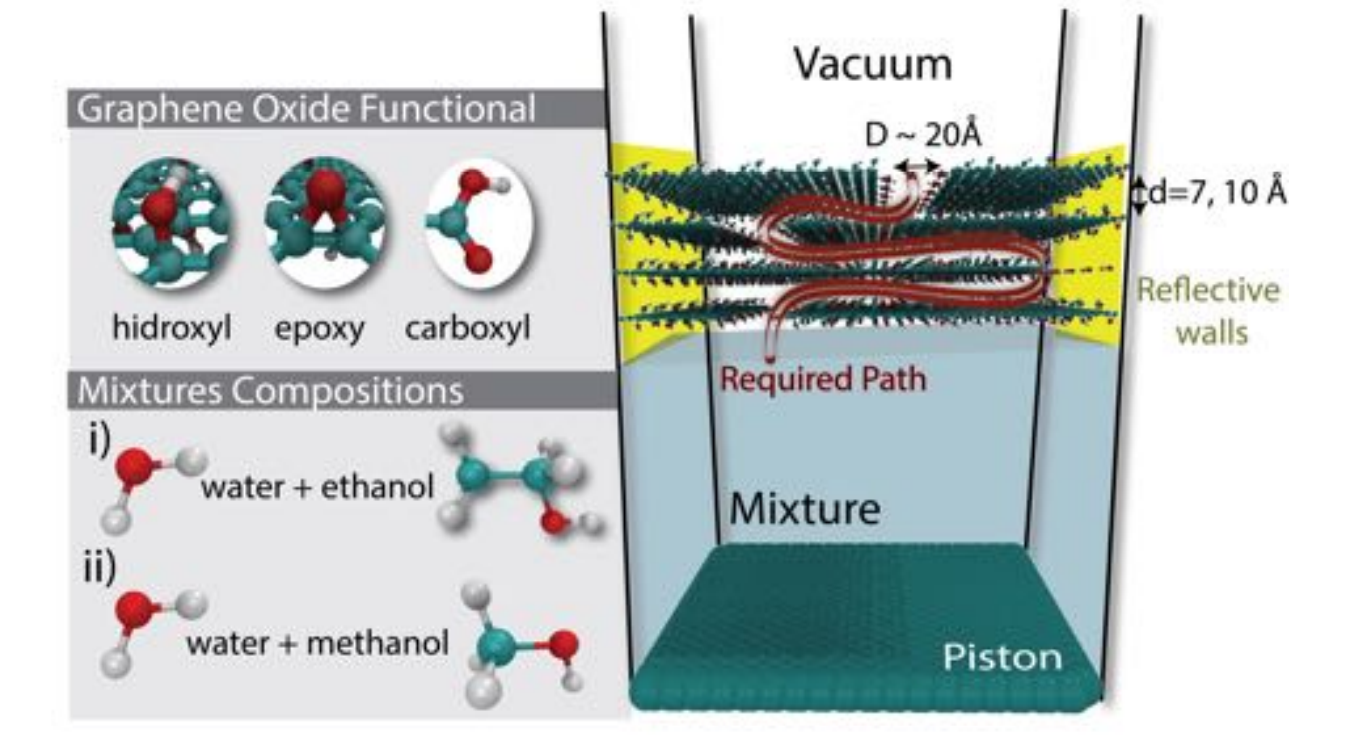
abstract = {Experimental evidences have shown that graphene oxide (GO) can be impermeable to liquids, vapors and gases, while it allows a fast permeation of water molecules. The understanding of filtration mechanisms came mostly from studies dedicated to water desalination, while very few works have been dedicated to distilling alcohols. In this work, we have investigated the molecular level mechanism underlying the alcohol/water separation inside GO membranes. A series of molecular dynamics and Grand-Canonical Monte Carlo simulations were carried out to probe the ethanol/water and methanol/water separation through GO membranes composed of multiple layered graphene-based sheets with different interlayer distance values and number of oxygen-containing functional groups. Our results show that the size exclusion and membrane affinities are not sufficient to explain the selectivity. Besides that, the favorable water molecular arrangement inside GO 2D-channels forming a robust H-bond network and the fast water diffusion are crucial for an effective separation mechanism. In other words, the separation phenomenon is not only governed by affinities with the membrane (enthalpic mechanisms) but mainly by the geometry and size factors (entropic mechanisms). We verified that the 2D geometry channel with optimal interlayer distance are key factors for designing more efficient alcohol-water separation membranes. Our findings are consistent with the available experimental data and contribute to clarify important aspects of the separation behavior of confined alcohol/water in GO membranes.},
note = {preprint arXiv:1702.00250},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Brunetto, Gustavo; Legoas, Sergio B; Coluci, Vitor R; Lucena, Liacir S; Galvao, Douglas S
Dynamics of Graphene Nanodrums Proceedings
Cambridge University Press, vol. 1284, 2011.
@proceedings{brunetto2011dynamics,
title = {Dynamics of Graphene Nanodrums},
author = {Brunetto, Gustavo and Legoas, Sergio B and Coluci, Vitor R and Lucena, Liacir S and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8195889&fileId=S1946427411002272},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1284},
pages = {mrsf10--1284},
publisher = {Cambridge University Press},
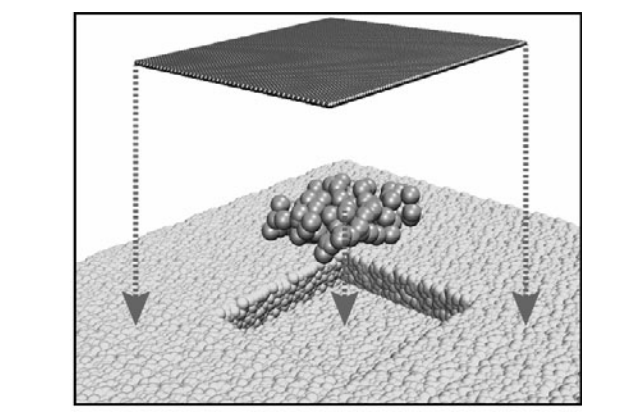
abstract = {Recently, it was proposed that graphene sheets deposited on silicon oxide can act as impermeable atomic membranes to standard gases, such as helium, argon, and nitrogen. It is assumed that graphene membrane is clamped over the surface due only to van der Waals forces. The leakage mechanism can be experimentally addressed only indirectly. In this work we have carried out molecular dynamics simulations to study this problem. We have considered nano-containers composed of a chamber of silicon oxide filled with gas and sealed by single and multi-layer graphene membranes. The obtained results are in good qualitative agreement with the experimental data. We observed that the graphene membranes remain attached to the substrate for pressure values up to two times the largest value experimentally investigated. We did not observe any gas leakage through the membrane/substrate interface until the critical limit is reached and then a sudden membrane detachment occurs.},
keywords = {},
pubstate = {published},
tppubtype = {proceedings}
}
2017
Borges, Daiane Damasceno; Woellner, Cristiano F; Autreto, Pedro AS; Galvao, Douglas S
2017, (preprint arXiv:1702.00250).
Abstract | Links | BibTeX | Tags: Filtration, Graphene Membranes, Molecular Dyanmics
@online{Borges2017b,
title = {Insights on the mechanism of water-alcohol separation in multilayer graphene oxide membranes: entropic versus enthalpic factors},
author = {Borges, Daiane Damasceno and Woellner, Cristiano F and Autreto, Pedro AS and Galvao, Douglas S},
url = {https://arxiv.org/abs/1706.06213},
year = {2017},
date = {2017-06-19},
abstract = {Experimental evidences have shown that graphene oxide (GO) can be impermeable to liquids, vapors and gases, while it allows a fast permeation of water molecules. The understanding of filtration mechanisms came mostly from studies dedicated to water desalination, while very few works have been dedicated to distilling alcohols. In this work, we have investigated the molecular level mechanism underlying the alcohol/water separation inside GO membranes. A series of molecular dynamics and Grand-Canonical Monte Carlo simulations were carried out to probe the ethanol/water and methanol/water separation through GO membranes composed of multiple layered graphene-based sheets with different interlayer distance values and number of oxygen-containing functional groups. Our results show that the size exclusion and membrane affinities are not sufficient to explain the selectivity. Besides that, the favorable water molecular arrangement inside GO 2D-channels forming a robust H-bond network and the fast water diffusion are crucial for an effective separation mechanism. In other words, the separation phenomenon is not only governed by affinities with the membrane (enthalpic mechanisms) but mainly by the geometry and size factors (entropic mechanisms). We verified that the 2D geometry channel with optimal interlayer distance are key factors for designing more efficient alcohol-water separation membranes. Our findings are consistent with the available experimental data and contribute to clarify important aspects of the separation behavior of confined alcohol/water in GO membranes.},
note = {preprint arXiv:1702.00250},
keywords = {Filtration, Graphene Membranes, Molecular Dyanmics},
pubstate = {published},
tppubtype = {online}
}
2011
Brunetto, Gustavo; Legoas, Sergio B; Coluci, Vitor R; Lucena, Liacir S; Galvao, Douglas S
Dynamics of Graphene Nanodrums Proceedings
Cambridge University Press, vol. 1284, 2011.
Abstract | Links | BibTeX | Tags: Graphene Membranes, Mechanical Properties, Nanodrum
@proceedings{brunetto2011dynamics,
title = {Dynamics of Graphene Nanodrums},
author = {Brunetto, Gustavo and Legoas, Sergio B and Coluci, Vitor R and Lucena, Liacir S and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8195889&fileId=S1946427411002272},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1284},
pages = {mrsf10--1284},
publisher = {Cambridge University Press},
abstract = {Recently, it was proposed that graphene sheets deposited on silicon oxide can act as impermeable atomic membranes to standard gases, such as helium, argon, and nitrogen. It is assumed that graphene membrane is clamped over the surface due only to van der Waals forces. The leakage mechanism can be experimentally addressed only indirectly. In this work we have carried out molecular dynamics simulations to study this problem. We have considered nano-containers composed of a chamber of silicon oxide filled with gas and sealed by single and multi-layer graphene membranes. The obtained results are in good qualitative agreement with the experimental data. We observed that the graphene membranes remain attached to the substrate for pressure values up to two times the largest value experimentally investigated. We did not observe any gas leakage through the membrane/substrate interface until the critical limit is reached and then a sudden membrane detachment occurs.},
keywords = {Graphene Membranes, Mechanical Properties, Nanodrum},
pubstate = {published},
tppubtype = {proceedings}
}
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ

