
Pedro Alves da Silva Autreto Cristiano Francisco Woellner, Douglas S. Galvao
One Side-Graphene Hydrogenation (Graphone): Substrate Effects Journal Article
In: MRS Advances, vol. 2016, 2016.
@article{Woellner2016b,
title = {One Side-Graphene Hydrogenation (Graphone): Substrate Effects},
author = {Cristiano Francisco Woellner, Pedro Alves da Silva Autreto, Douglas S. Galvao},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=10234793&fulltextType=RA&fileId=S2059852116001961},
doi = {DOI: 10.1557/adv.2016.196},
year = {2016},
date = {2016-03-01},
journal = {MRS Advances},
volume = {2016},
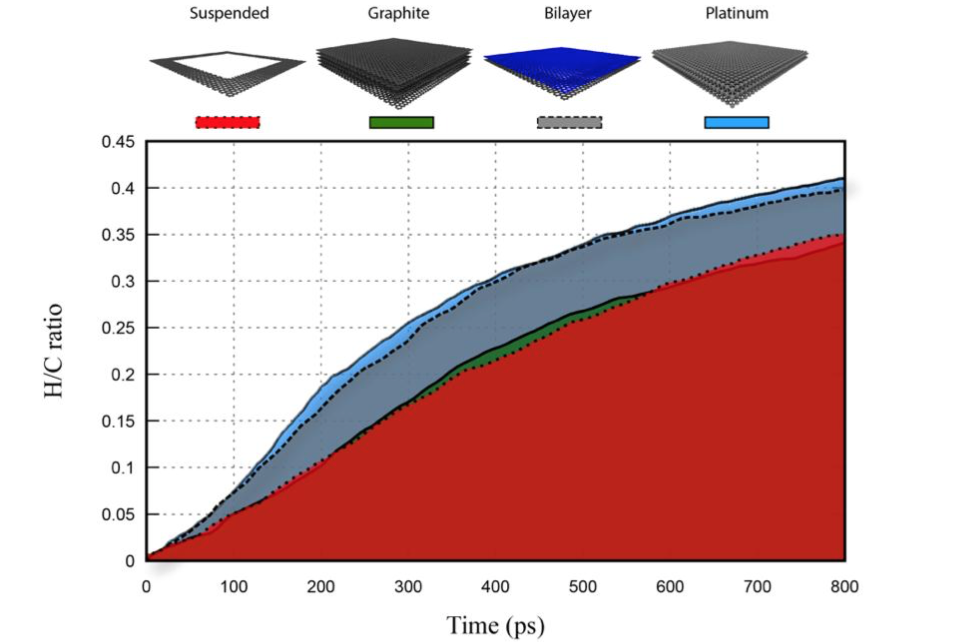
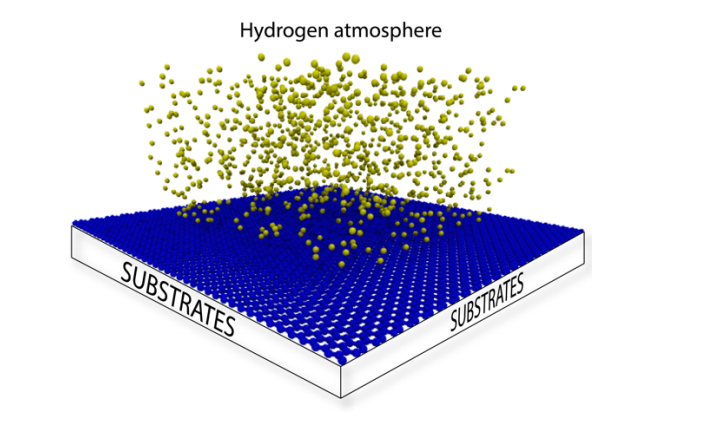
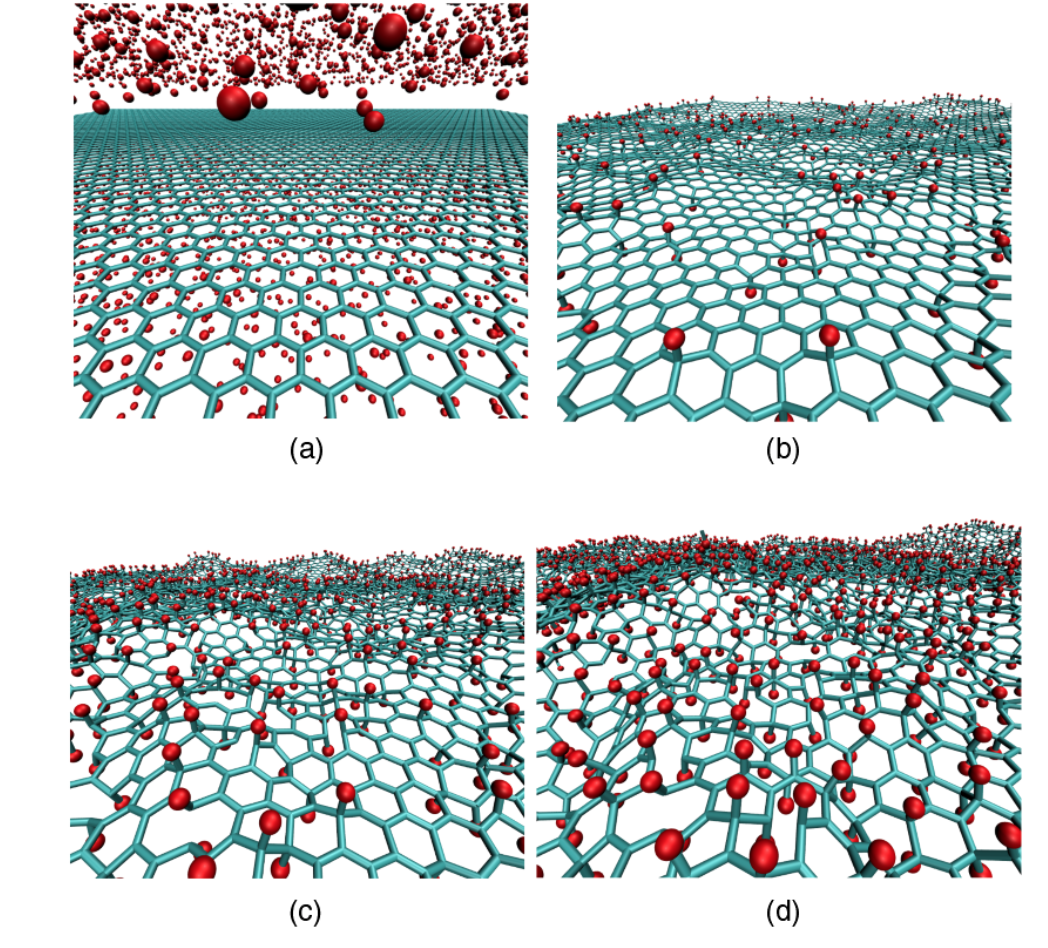
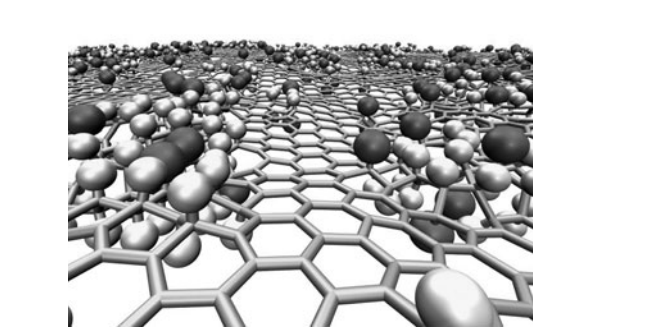
abstract = {Recent studies on graphene hydrogenation processes showed that hydrogenation occurs via island growing domains, however how the substrate can affect the hydrogenation dynamics and/or pattern formation has not been yet properly investigated. In this work we have addressed these issues through fully atomistic reactive molecular dynamics simulations. We investigated the structural and dynamical aspects of the hydrogenation of graphene membranes (one-side hydrogenation, the so called graphone structure) on different substrates (graphene, few-layers graphene, graphite and platinum). Our results also show that the observed hydrogenation rates are very sensitive to the substrate type. For all investigated cases, the largest fraction of hydrogenated carbon atoms was for platinum substrates. Our results also show that a significant number of randomly distributed H clusters are formed during the early stages of the hydrogenation process, regardless of the type of substrate. These results suggest that, similarly to graphane formation, large perfect graphone-like domains are unlikely to be formed. These findings are especially important since experiments have showed that cluster formation influences the electronic transport properties in hydrogenated graphene.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Woellner, Cristiano Francisco; Autreto, Pedro Alves da Silva; Galvao, Douglas S
One Side-Graphene Hydrogenation (Graphone): Substrate Effects Online
2016, visited: 18.01.2016, ((ArXiv preprint)).
@online{Woellner2016,
title = {One Side-Graphene Hydrogenation (Graphone): Substrate Effects},
author = {Woellner, Cristiano Francisco and Autreto, Pedro Alves da Silva and Galvao, Douglas S},
url = {http://arxiv.org/abs/1601.04484},
year = {2016},
date = {2016-01-18},
urldate = {2016-01-18},
abstract = {Recent studies on graphene hydrogenation processes showed that hydrogenation occurs
via island growing domains, however how the substrate can affect the hydrogenation dynamics
and/or pattern formation has not been yet properly investigated. In this work we have addressed
these issues through fully atomistic reactive molecular dynamics simulations. We investigated
the structural and dynamical aspects of the hydrogenation of graphene membranes (one-side
hydrogenation, the so called graphone structure) on different substrates (graphene, few-layers
graphene, graphite and platinum). Our results also show that the observed hydrogenation rates
are very sensitive to the substrate type. For all investigated cases, the largest fraction of
hydrogenated carbon atoms was for platinum substrates. Our results also show that a significant
number of randomly distributed H clusters are formed during the early stages of the
hydrogenation process, regardless of the type of substrate and temperature. These results suggest
that, similarly to graphane formation, large perfect graphone-like domains are unlikely to be
formed. These findings are especially important since experiments have showed that cluster
formation influences the electronic transport properties in hydrogenated graphene.},
note = {(ArXiv preprint)},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
via island growing domains, however how the substrate can affect the hydrogenation dynamics
and/or pattern formation has not been yet properly investigated. In this work we have addressed
these issues through fully atomistic reactive molecular dynamics simulations. We investigated
the structural and dynamical aspects of the hydrogenation of graphene membranes (one-side
hydrogenation, the so called graphone structure) on different substrates (graphene, few-layers
graphene, graphite and platinum). Our results also show that the observed hydrogenation rates
are very sensitive to the substrate type. For all investigated cases, the largest fraction of
hydrogenated carbon atoms was for platinum substrates. Our results also show that a significant
number of randomly distributed H clusters are formed during the early stages of the
hydrogenation process, regardless of the type of substrate and temperature. These results suggest
that, similarly to graphane formation, large perfect graphone-like domains are unlikely to be
formed. These findings are especially important since experiments have showed that cluster
formation influences the electronic transport properties in hydrogenated graphene.
Paupitz, R; Autreto, Pedro AS; Legoas, SB; Srinivasan, S Goverapet; van Duin, Adri CT; Galvao, DS
Graphene to fluorographene and fluorographane: a theoretical study Journal Article
In: Nanotechnology, vol. 24, no. 3, pp. 035706, 2013.
@article{paupitz2013graphene,
title = {Graphene to fluorographene and fluorographane: a theoretical study},
author = {Paupitz, R and Autreto, Pedro AS and Legoas, SB and Srinivasan, S Goverapet and van Duin, Adri CT and Galvao, DS},
url = {http://iopscience.iop.org/0957-4484/24/3/035706},
year = {2013},
date = {2013-01-01},
journal = {Nanotechnology},
volume = {24},
number = {3},
pages = {035706},
publisher = {IOP Publishing},
abstract = {We report here a fully reactive molecular dynamics study on the structural and dynamical aspects of the fluorination of graphene membranes (fluorographene). Our results show that fluorination tends to produce defective areas on the graphene membranes with significant distortions of carbon–carbon bonds. Depending on the amount of incorporated fluorine atoms, large membrane holes were observed due to carbon atom losses. These results may explain the broad distribution of the structural lattice parameter values experimentally observed. We have also investigated the effects of mixing hydrogen and fluorine atoms on the graphene functionalization. Our results show that, when in small amounts, the presence of hydrogen atoms produces a significant decrease in the rate of fluorine incorporation onto the membrane. On the other hand, when fluorine is the minority element, it produces a significant catalytic effect on the rate of hydrogen incorporation. We have also observed the spontaneous formation of new hybrid structures with different stable configurations (chair-like, zigzag-like and boat-like) which we named fluorographane.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Autreto, Pedro AS; Flores, Marcelo Z; Legoas, Sergio B; Santos, Ricardo PB; Galvao, Douglas S
Cambridge University Press, vol. 1284, 2011.
@proceedings{autreto2011fully,
title = {A Fully Atomistic Reactive Molecular Dynamics Study on the Formation of Graphane from Graphene Hydrogenated Membranes.},
author = {Autreto, Pedro AS and Flores, Marcelo Z and Legoas, Sergio B and Santos, Ricardo PB and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8364784&fileId=S1946427411013583},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1284},
pages = {mrsf10--1284},
publisher = {Cambridge University Press},
abstract = {Using fully reactive molecular dynamics methodologies we investigated the structural and dynamical aspects of the fluorination mechanism leading to fluorographene formation from graphene membranes. Fluorination tends to produce significant defective areas on the membranes with variation on the typical carbon-carbon distances, sometimes with the presence of large holes due to carbon losses. The results obtained in our simulations are in good agreement with the broad distribution of values for the lattice parameter experimentally observed. We have also investigated mixed atmospheres composed by H and F atoms. When H is present in small quantities an expressive reduction on the rate of incorporation of fluorine was observed. On the other hand when fluorine atoms are present in small quantities in a hydrogen atmosphere, they induce an increasing on the hydrogen incorporation and the formation of locally self-organized structure of adsorbed H and F atoms.},
keywords = {},
pubstate = {published},
tppubtype = {proceedings}
}
Santos, Ricardo PB; Autreto, Pedro AS; Legoas, Sergio B; Galvao, Douglas S
Cambridge University Press, vol. 1344, 2011.
@proceedings{santos2011dynamics,
title = {The Dynamics of Formation of Graphane-like Fluorinated Graphene Membranes (Fluorographene): A Reactive Molecular Dynamics Study},
author = {Santos, Ricardo PB and Autreto, Pedro AS and Legoas, Sergio B and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayFulltext?type=1&fid=8237871&jid=OPL&volumeId=1284&issueId=-1&aid=8237869},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1344},
pages = {mrss11--1344},
publisher = {Cambridge University Press},
abstract = {Recently, Elias et al. (Science 323, 610 (2009).) reported the experimental realization of
the formation of graphane from hydrogenation of graphene membranes under cold plasma
exposure. In graphane, the carbon-carbon bonds are in sp3
configuration, as opposed to the sp2
hybridization of graphene, and the C–H bonds exhibit an alternating pattern (up and down with
relation to the plane defined by the carbon atoms). In this work we have investigated, using
reactive molecular dynamics simulations, the role of H frustration (breaking the H atoms up and
down alternating pattern) in graphane-like structures. Our results show that a significant
percentage of uncorrelated H frustrated domains are formed in the early stages of the
hydrogenation process, leading to membrane shrinkage and extensive membrane corrugations.
This might explain the significant broad distribution of values of lattice parameter
experimentally observed. For comparison purposes we have also analyzed fluorinated graphanelike
structures. Our results show that similarly to H, F atoms also create significant uncorrelated
frustrated domains on graphene membranes. },
keywords = {},
pubstate = {published},
tppubtype = {proceedings}
}
the formation of graphane from hydrogenation of graphene membranes under cold plasma
exposure. In graphane, the carbon-carbon bonds are in sp3
configuration, as opposed to the sp2
hybridization of graphene, and the C–H bonds exhibit an alternating pattern (up and down with
relation to the plane defined by the carbon atoms). In this work we have investigated, using
reactive molecular dynamics simulations, the role of H frustration (breaking the H atoms up and
down alternating pattern) in graphane-like structures. Our results show that a significant
percentage of uncorrelated H frustrated domains are formed in the early stages of the
hydrogenation process, leading to membrane shrinkage and extensive membrane corrugations.
This might explain the significant broad distribution of values of lattice parameter
experimentally observed. For comparison purposes we have also analyzed fluorinated graphanelike
structures. Our results show that similarly to H, F atoms also create significant uncorrelated
frustrated domains on graphene membranes.
Legoas, Sergio B; Autreto, Pedro AS; Flores, Marcelo ZS; Galvao, Douglas S
Graphene to graphane: the role of H frustration in lattice contraction Journal Article
In: arXiv preprint arXiv:0903.0278, 2009.
@article{legoas2009graphene,
title = {Graphene to graphane: the role of H frustration in lattice contraction},
author = {Legoas, Sergio B and Autreto, Pedro AS and Flores, Marcelo ZS and Galvao, Douglas S},
url = {http://arxiv.org/abs/0903.0278},
year = {2009},
date = {2009-01-01},
journal = {arXiv preprint arXiv:0903.0278},
abstract = {Graphane is a two-dimensional system consisting of a single planar layer of fully saturated (sp3 hybridization) carbon atoms with H atoms attached to them in an alternating pattern (up and down with relation to the plane defined by the carbon atoms). Stable graphane structures were theoretically predicted to exist some years ago and just experimentally realized through hydrogenation of graphene membranes. In this work we have investigated using textit{ab initio} and reactive molecular dynamics the role of H frustration (breaking the H atoms up and down alternating pattern) in graphane-like structures. Our results show that H frustration significantly contributes to lattice contraction. The dynamical aspects of converting graphene to graphane is also addressed.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2016
Pedro Alves da Silva Autreto Cristiano Francisco Woellner, Douglas S. Galvao
One Side-Graphene Hydrogenation (Graphone): Substrate Effects Journal Article
In: MRS Advances, vol. 2016, 2016.
Abstract | Links | BibTeX | Tags: Graphane, Graphene, graphone, Molecular Dynamics
@article{Woellner2016b,
title = {One Side-Graphene Hydrogenation (Graphone): Substrate Effects},
author = {Cristiano Francisco Woellner, Pedro Alves da Silva Autreto, Douglas S. Galvao},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=10234793&fulltextType=RA&fileId=S2059852116001961},
doi = {DOI: 10.1557/adv.2016.196},
year = {2016},
date = {2016-03-01},
journal = {MRS Advances},
volume = {2016},
abstract = {Recent studies on graphene hydrogenation processes showed that hydrogenation occurs via island growing domains, however how the substrate can affect the hydrogenation dynamics and/or pattern formation has not been yet properly investigated. In this work we have addressed these issues through fully atomistic reactive molecular dynamics simulations. We investigated the structural and dynamical aspects of the hydrogenation of graphene membranes (one-side hydrogenation, the so called graphone structure) on different substrates (graphene, few-layers graphene, graphite and platinum). Our results also show that the observed hydrogenation rates are very sensitive to the substrate type. For all investigated cases, the largest fraction of hydrogenated carbon atoms was for platinum substrates. Our results also show that a significant number of randomly distributed H clusters are formed during the early stages of the hydrogenation process, regardless of the type of substrate. These results suggest that, similarly to graphane formation, large perfect graphone-like domains are unlikely to be formed. These findings are especially important since experiments have showed that cluster formation influences the electronic transport properties in hydrogenated graphene.
},
keywords = {Graphane, Graphene, graphone, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Woellner, Cristiano Francisco; Autreto, Pedro Alves da Silva; Galvao, Douglas S
One Side-Graphene Hydrogenation (Graphone): Substrate Effects Online
2016, visited: 18.01.2016, ((ArXiv preprint)).
Abstract | Links | BibTeX | Tags: Graphane, Graphene, graphone, Molecular Dynamics
@online{Woellner2016,
title = {One Side-Graphene Hydrogenation (Graphone): Substrate Effects},
author = {Woellner, Cristiano Francisco and Autreto, Pedro Alves da Silva and Galvao, Douglas S},
url = {http://arxiv.org/abs/1601.04484},
year = {2016},
date = {2016-01-18},
urldate = {2016-01-18},
abstract = {Recent studies on graphene hydrogenation processes showed that hydrogenation occurs
via island growing domains, however how the substrate can affect the hydrogenation dynamics
and/or pattern formation has not been yet properly investigated. In this work we have addressed
these issues through fully atomistic reactive molecular dynamics simulations. We investigated
the structural and dynamical aspects of the hydrogenation of graphene membranes (one-side
hydrogenation, the so called graphone structure) on different substrates (graphene, few-layers
graphene, graphite and platinum). Our results also show that the observed hydrogenation rates
are very sensitive to the substrate type. For all investigated cases, the largest fraction of
hydrogenated carbon atoms was for platinum substrates. Our results also show that a significant
number of randomly distributed H clusters are formed during the early stages of the
hydrogenation process, regardless of the type of substrate and temperature. These results suggest
that, similarly to graphane formation, large perfect graphone-like domains are unlikely to be
formed. These findings are especially important since experiments have showed that cluster
formation influences the electronic transport properties in hydrogenated graphene.},
note = {(ArXiv preprint)},
keywords = {Graphane, Graphene, graphone, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}
via island growing domains, however how the substrate can affect the hydrogenation dynamics
and/or pattern formation has not been yet properly investigated. In this work we have addressed
these issues through fully atomistic reactive molecular dynamics simulations. We investigated
the structural and dynamical aspects of the hydrogenation of graphene membranes (one-side
hydrogenation, the so called graphone structure) on different substrates (graphene, few-layers
graphene, graphite and platinum). Our results also show that the observed hydrogenation rates
are very sensitive to the substrate type. For all investigated cases, the largest fraction of
hydrogenated carbon atoms was for platinum substrates. Our results also show that a significant
number of randomly distributed H clusters are formed during the early stages of the
hydrogenation process, regardless of the type of substrate and temperature. These results suggest
that, similarly to graphane formation, large perfect graphone-like domains are unlikely to be
formed. These findings are especially important since experiments have showed that cluster
formation influences the electronic transport properties in hydrogenated graphene.
2013
Paupitz, R; Autreto, Pedro AS; Legoas, SB; Srinivasan, S Goverapet; van Duin, Adri CT; Galvao, DS
Graphene to fluorographene and fluorographane: a theoretical study Journal Article
In: Nanotechnology, vol. 24, no. 3, pp. 035706, 2013.
Abstract | Links | BibTeX | Tags: Fluorographene, Graphane, Graphene
@article{paupitz2013graphene,
title = {Graphene to fluorographene and fluorographane: a theoretical study},
author = {Paupitz, R and Autreto, Pedro AS and Legoas, SB and Srinivasan, S Goverapet and van Duin, Adri CT and Galvao, DS},
url = {http://iopscience.iop.org/0957-4484/24/3/035706},
year = {2013},
date = {2013-01-01},
journal = {Nanotechnology},
volume = {24},
number = {3},
pages = {035706},
publisher = {IOP Publishing},
abstract = {We report here a fully reactive molecular dynamics study on the structural and dynamical aspects of the fluorination of graphene membranes (fluorographene). Our results show that fluorination tends to produce defective areas on the graphene membranes with significant distortions of carbon–carbon bonds. Depending on the amount of incorporated fluorine atoms, large membrane holes were observed due to carbon atom losses. These results may explain the broad distribution of the structural lattice parameter values experimentally observed. We have also investigated the effects of mixing hydrogen and fluorine atoms on the graphene functionalization. Our results show that, when in small amounts, the presence of hydrogen atoms produces a significant decrease in the rate of fluorine incorporation onto the membrane. On the other hand, when fluorine is the minority element, it produces a significant catalytic effect on the rate of hydrogen incorporation. We have also observed the spontaneous formation of new hybrid structures with different stable configurations (chair-like, zigzag-like and boat-like) which we named fluorographane.},
keywords = {Fluorographene, Graphane, Graphene},
pubstate = {published},
tppubtype = {article}
}
2011
Autreto, Pedro AS; Flores, Marcelo Z; Legoas, Sergio B; Santos, Ricardo PB; Galvao, Douglas S
Cambridge University Press, vol. 1284, 2011.
Abstract | Links | BibTeX | Tags: Graphane, Graphene, Hydrogenation, Molecular Dynamics
@proceedings{autreto2011fully,
title = {A Fully Atomistic Reactive Molecular Dynamics Study on the Formation of Graphane from Graphene Hydrogenated Membranes.},
author = {Autreto, Pedro AS and Flores, Marcelo Z and Legoas, Sergio B and Santos, Ricardo PB and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8364784&fileId=S1946427411013583},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1284},
pages = {mrsf10--1284},
publisher = {Cambridge University Press},
abstract = {Using fully reactive molecular dynamics methodologies we investigated the structural and dynamical aspects of the fluorination mechanism leading to fluorographene formation from graphene membranes. Fluorination tends to produce significant defective areas on the membranes with variation on the typical carbon-carbon distances, sometimes with the presence of large holes due to carbon losses. The results obtained in our simulations are in good agreement with the broad distribution of values for the lattice parameter experimentally observed. We have also investigated mixed atmospheres composed by H and F atoms. When H is present in small quantities an expressive reduction on the rate of incorporation of fluorine was observed. On the other hand when fluorine atoms are present in small quantities in a hydrogen atmosphere, they induce an increasing on the hydrogen incorporation and the formation of locally self-organized structure of adsorbed H and F atoms.},
keywords = {Graphane, Graphene, Hydrogenation, Molecular Dynamics},
pubstate = {published},
tppubtype = {proceedings}
}
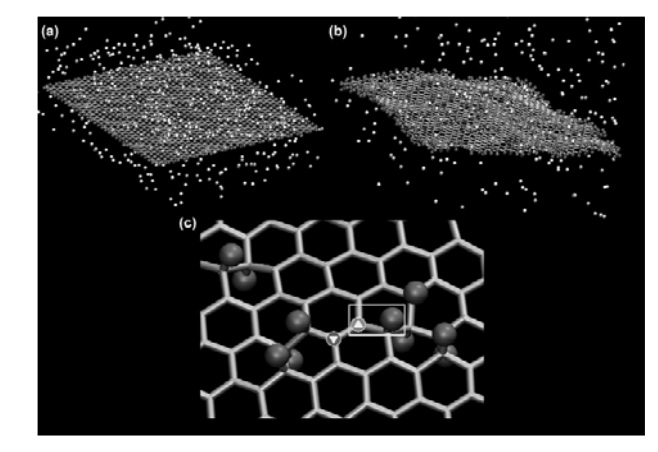
Santos, Ricardo PB; Autreto, Pedro AS; Legoas, Sergio B; Galvao, Douglas S
Cambridge University Press, vol. 1344, 2011.
Abstract | Links | BibTeX | Tags: Fluorographene, Functionalization, Graphane, Graphene
@proceedings{santos2011dynamics,
title = {The Dynamics of Formation of Graphane-like Fluorinated Graphene Membranes (Fluorographene): A Reactive Molecular Dynamics Study},
author = {Santos, Ricardo PB and Autreto, Pedro AS and Legoas, Sergio B and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayFulltext?type=1&fid=8237871&jid=OPL&volumeId=1284&issueId=-1&aid=8237869},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1344},
pages = {mrss11--1344},
publisher = {Cambridge University Press},
abstract = {Recently, Elias et al. (Science 323, 610 (2009).) reported the experimental realization of
the formation of graphane from hydrogenation of graphene membranes under cold plasma
exposure. In graphane, the carbon-carbon bonds are in sp3
configuration, as opposed to the sp2
hybridization of graphene, and the C–H bonds exhibit an alternating pattern (up and down with
relation to the plane defined by the carbon atoms). In this work we have investigated, using
reactive molecular dynamics simulations, the role of H frustration (breaking the H atoms up and
down alternating pattern) in graphane-like structures. Our results show that a significant
percentage of uncorrelated H frustrated domains are formed in the early stages of the
hydrogenation process, leading to membrane shrinkage and extensive membrane corrugations.
This might explain the significant broad distribution of values of lattice parameter
experimentally observed. For comparison purposes we have also analyzed fluorinated graphanelike
structures. Our results show that similarly to H, F atoms also create significant uncorrelated
frustrated domains on graphene membranes. },
keywords = {Fluorographene, Functionalization, Graphane, Graphene},
pubstate = {published},
tppubtype = {proceedings}
}
the formation of graphane from hydrogenation of graphene membranes under cold plasma
exposure. In graphane, the carbon-carbon bonds are in sp3
configuration, as opposed to the sp2
hybridization of graphene, and the C–H bonds exhibit an alternating pattern (up and down with
relation to the plane defined by the carbon atoms). In this work we have investigated, using
reactive molecular dynamics simulations, the role of H frustration (breaking the H atoms up and
down alternating pattern) in graphane-like structures. Our results show that a significant
percentage of uncorrelated H frustrated domains are formed in the early stages of the
hydrogenation process, leading to membrane shrinkage and extensive membrane corrugations.
This might explain the significant broad distribution of values of lattice parameter
experimentally observed. For comparison purposes we have also analyzed fluorinated graphanelike
structures. Our results show that similarly to H, F atoms also create significant uncorrelated
frustrated domains on graphene membranes.
2009
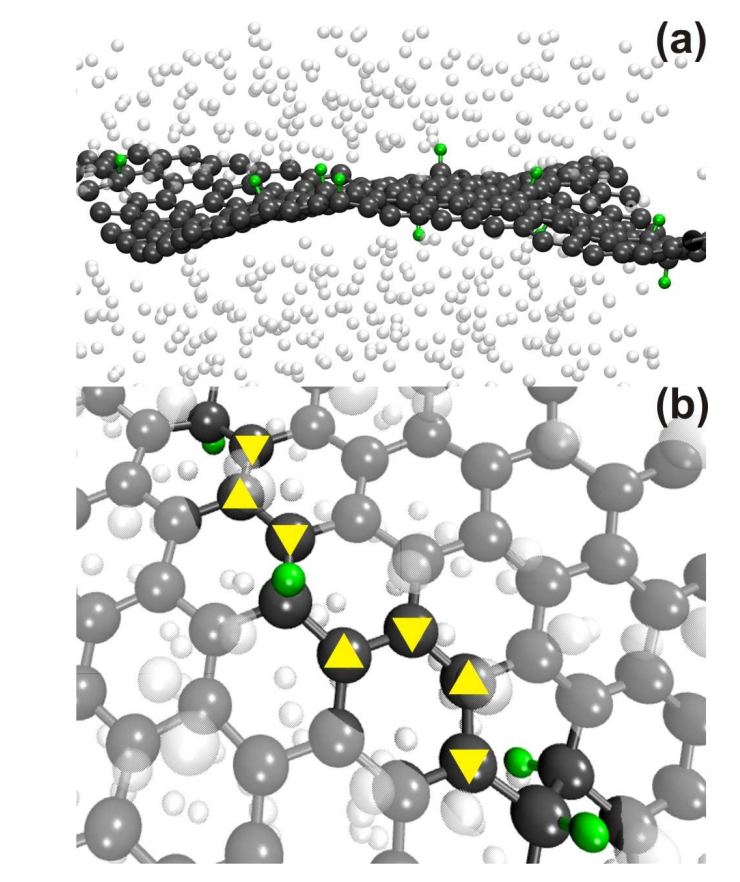
Legoas, Sergio B; Autreto, Pedro AS; Flores, Marcelo ZS; Galvao, Douglas S
Graphene to graphane: the role of H frustration in lattice contraction Journal Article
In: arXiv preprint arXiv:0903.0278, 2009.
Abstract | Links | BibTeX | Tags: Functionalization, Graphane, Graphene, Hydrogenation
@article{legoas2009graphene,
title = {Graphene to graphane: the role of H frustration in lattice contraction},
author = {Legoas, Sergio B and Autreto, Pedro AS and Flores, Marcelo ZS and Galvao, Douglas S},
url = {http://arxiv.org/abs/0903.0278},
year = {2009},
date = {2009-01-01},
journal = {arXiv preprint arXiv:0903.0278},
abstract = {Graphane is a two-dimensional system consisting of a single planar layer of fully saturated (sp3 hybridization) carbon atoms with H atoms attached to them in an alternating pattern (up and down with relation to the plane defined by the carbon atoms). Stable graphane structures were theoretically predicted to exist some years ago and just experimentally realized through hydrogenation of graphene membranes. In this work we have investigated using textit{ab initio} and reactive molecular dynamics the role of H frustration (breaking the H atoms up and down alternating pattern) in graphane-like structures. Our results show that H frustration significantly contributes to lattice contraction. The dynamical aspects of converting graphene to graphane is also addressed.},
keywords = {Functionalization, Graphane, Graphene, Hydrogenation},
pubstate = {published},
tppubtype = {article}
}
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ

