
Borges, Daiane D; Woellner, Cristiano F; Autreto, Pedro AS; Galvao, Douglas S
2017, (preprint arXiv:1702.00250).
@online{Borges2017,
title = {Water Permeation through Layered Graphene-based Membranes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Borges, Daiane D and Woellner, Cristiano F and Autreto, Pedro AS and Galvao, Douglas S},
url = {https://arxiv.org/abs/1702.00250},
year = {2017},
date = {2017-02-01},
abstract = {Graphene-based membranes have been investigated as promising candidates for water
filtration and gas separation applications. Experimental evidences have shown that graphene
oxide can be impermeable to liquids, vapors and gases, while allowing a fast permeation of water
molecules. This phenomenon has been attributed to the formation of a network of nano
capillaries that allow nearly frictionless water flow while blocking other molecules by steric
hindrance effects. It is supposed that water molecules are transported through the percolated twodimensional
channels formed between graphene-based sheets. Although these channels allow
fast water permeation in such materials, the flow rates are strongly dependent on how the
membranes are fabricated. Also, some fundamental issues regarding the nanoscale mechanisms
of water permeation are still not fully understood and their interpretation remains controversial.
In this work, we have investigated the dynamics of water permeation through pristine graphene
and graphene oxide model membranes. We have carried out fully atomistic classical molecular
dynamics simulations of systems composed of multiple layered graphene-based sheets into
contact with a water reservoir under controlled thermodynamics conditions (e. g., by varying
temperature and pressure values). We have systematically analyzed how the transport dynamics
of the confined nanofluids depend on the interlayer distances and the role of the oxide functional
groups. Our results show the water flux is much more effective for graphene than for graphene
oxide membranes. These results are attributed to the H-bonds formation between oxide
functional groups and water, which traps the water molecules and precludes ultrafast water
transport through the nanochannels.},
note = {preprint arXiv:1702.00250},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
filtration and gas separation applications. Experimental evidences have shown that graphene
oxide can be impermeable to liquids, vapors and gases, while allowing a fast permeation of water
molecules. This phenomenon has been attributed to the formation of a network of nano
capillaries that allow nearly frictionless water flow while blocking other molecules by steric
hindrance effects. It is supposed that water molecules are transported through the percolated twodimensional
channels formed between graphene-based sheets. Although these channels allow
fast water permeation in such materials, the flow rates are strongly dependent on how the
membranes are fabricated. Also, some fundamental issues regarding the nanoscale mechanisms
of water permeation are still not fully understood and their interpretation remains controversial.
In this work, we have investigated the dynamics of water permeation through pristine graphene
and graphene oxide model membranes. We have carried out fully atomistic classical molecular
dynamics simulations of systems composed of multiple layered graphene-based sheets into
contact with a water reservoir under controlled thermodynamics conditions (e. g., by varying
temperature and pressure values). We have systematically analyzed how the transport dynamics
of the confined nanofluids depend on the interlayer distances and the role of the oxide functional
groups. Our results show the water flux is much more effective for graphene than for graphene
oxide membranes. These results are attributed to the H-bonds formation between oxide
functional groups and water, which traps the water molecules and precludes ultrafast water
transport through the nanochannels.
Solis, Daniel; Woellner, Cristiano F; Borges, Daiane D; Galvao, Douglas S
Mechanical and Thermal Stability of Graphyne and Graphdiyne Nanoscrolls Journal Article
In: MRS Advances, vol. 2017, pp. 129-134, 2017.
@article{Solis2017,
title = {Mechanical and Thermal Stability of Graphyne and Graphdiyne Nanoscrolls},
author = {Solis, Daniel and Woellner, Cristiano F and Borges, Daiane D and Galvao, Douglas S},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/mechanical-and-thermal-stability-of-graphyne-and-graphdiyne-nanoscrolls/202E7B7C471411200DE9D05C264726B8},
doi = {10.1557/adv.2017.130},
year = {2017},
date = {2017-02-01},
journal = {MRS Advances},
volume = {2017},
pages = {129-134},
abstract = {Graphynes and graphdiynes are carbon 2D allotrope structures presenting both sp2 and sp hybridized atoms. These materials have been theoretically predicted but due to intrinsic difficulties in their synthesis, only recently some of these structures have been experimentally realized. Graphyne nanoscrolls are structures obtained by rolling up graphyne sheets into papyrus-like structures. In this work, we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics of nanoscroll formation for a series of graphyne (α, β, and δ types) structures. We have also investigated their thermal stability for a temperature range of 200-1000K. Our results show that stable nanoscrolls can be formed for all structures considered here. Their stability depends on a critical value of the ratio between length and height of the graphyne sheets. Our findings also show that these structures are structurally less stable then graphene-based nanoscrolls. This can be explained by the graphyne higher structural porosity which results in a decreased pi-pi stacking interactions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Jaques, Ygor M; Galvao, Douglas S
Permeation of Water Nanodroplets on Carbon Nanotubes Forests Journal Article
In: MRS Advances, vol. 2017, pp. 123-128, 2017.
@article{Jaques2017b,
title = {Permeation of Water Nanodroplets on Carbon Nanotubes Forests},
author = {Jaques, Ygor M and Galvao, Douglas S},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/permeation-of-water-nanodroplets-on-carbon-nanotubes-forests/99C67F3DC0AD10DB1A4580CC8CEFDF58},
doi = {10.1557/adv.2017.129},
year = {2017},
date = {2017-01-31},
journal = {MRS Advances},
volume = {2017},
pages = {123-128},
abstract = {Fully atomistic molecular dynamics simulations were carried out to investigate how a liquid-like water droplet behaves when into contact with a nanopore formed by carbon nanotube arrays. We have considered different tube arrays, varying the spacing between them, as well as, different chemical functionalizations on the uncapped nanotubes. Our results show that simple functionalizations (for instance, hydrogen ones) allow tuning up the wetting surface properties increasing the permeation of liquid inside the nanopore. For functionalizations that increase the surface hydrophilicity, even when the pore size is significantly increased the droplet remains at the surface without tube permeation.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Jaques, Ygor M; Galvao, Douglas S
Nanodroplets Behavior on Graphdiyne Membranes Journal Article
In: MRS Advances, vol. 2017, pp. 1-6, 2017.
@article{Jaques2017,
title = {Nanodroplets Behavior on Graphdiyne Membranes},
author = {Jaques, Ygor M and Galvao, Douglas S},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/nanodroplets-behavior-on-graphdiyne-membranes/16AD56CAD07570E7F4F194A56E9680C3},
doi = {10.1557/adv.2017.128},
year = {2017},
date = {2017-01-30},
journal = {MRS Advances},
volume = {2017},
pages = {1-6},
abstract = {In this work we have investigated, by fully atomistic reactive (force field ReaxFF) molecular dynamics simulations, some aspects of impact dynamics of water nanodroplets on graphdiyne-like membranes. We simulated graphdiyne-supported membranes impacted by nanodroplets at different velocities (from 100 up to 1500 m/s). The results show that due to the graphdiyne porous and elastic structure, the droplets present an impact dynamics very complex in relation to the ones observed for graphene membranes. Under impact the droplets spread over the surface with a maximum contact radius proportional to the impact velocity. Depending on the energy impact value, a number of water molecules were able to percolate the nanopore sheets. However, even in these cases the droplet shape is preserved and the main differences between the different impact velocities cases reside on the splashing pattern at the maximum spreading.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Bizao, Rafael A; Machado, Leonardo D; de Sousa, Jose M; Pugno, Nicola M; Galvao, Douglas S
Scale Effects on the Ballistic Penetration of Graphene Sheets Online
2017, (preprint arXiv:1701.07367).
@online{Bizao2017c,
title = {Scale Effects on the Ballistic Penetration of Graphene Sheets},
author = {Bizao, Rafael A and Machado, Leonardo D and de Sousa, Jose M and Pugno, Nicola M and Galvao, Douglas S},
url = {https://arxiv.org/pdf/1701.07367.pdf},
year = {2017},
date = {2017-01-25},
abstract = {Carbon nanostructures are promising ballistic protection materials,
due to their low density and excellent mechanical properties. Recent
experimental and computational investigations on the behavior
of graphene under impact conditions revealed exceptional energy absorption
properties as well. However, the reported numerical and experimental
values differ by an order of magnitude. In this work, we
combined numerical and analytical modeling to address this issue. In
the numerical part, we employed reactive molecular dynamics to carry
out ballistic tests on single and double-layered graphene sheets. We
used velocity values within the range tested in experiments. Our numerical
and the experimental results were used to determine parameters
for a scaling law, which is in good agreement with all experimental
and simulation results. We find that the specific penetration energy
decreases as the number of layers (N) increases, from ∼ 25 MJ/kg for
N = 1 to ∼ 0.26 MJ/kg as N → ∞. These scale effects explain the
apparent discrepancy between simulations and experiments.},
note = {preprint arXiv:1701.07367},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
due to their low density and excellent mechanical properties. Recent
experimental and computational investigations on the behavior
of graphene under impact conditions revealed exceptional energy absorption
properties as well. However, the reported numerical and experimental
values differ by an order of magnitude. In this work, we
combined numerical and analytical modeling to address this issue. In
the numerical part, we employed reactive molecular dynamics to carry
out ballistic tests on single and double-layered graphene sheets. We
used velocity values within the range tested in experiments. Our numerical
and the experimental results were used to determine parameters
for a scaling law, which is in good agreement with all experimental
and simulation results. We find that the specific penetration energy
decreases as the number of layers (N) increases, from ∼ 25 MJ/kg for
N = 1 to ∼ 0.26 MJ/kg as N → ∞. These scale effects explain the
apparent discrepancy between simulations and experiments.
Peter Samora Owuor Alin Cristian Chipara, Sanjit Bhowmick
Structural Reinforcement through Liquid Encapsulation Journal Article
In: Advanced Materials Interfaces, vol. 4, pp. 1600781, 2017.
@article{Chipara2017,
title = {Structural Reinforcement through Liquid Encapsulation},
author = {Alin Cristian Chipara, Peter Samora Owuor, Sanjit Bhowmick, Gustavo Brunetto, SA Asif, Mircea Chipara, Robert Vajtai, Jun Lou, Douglas S Galvao, Chandra Sekhar Tiwary, Pulickel M Ajayan},
url = {http://onlinelibrary.wiley.com/doi/10.1002/admi.201600781/full},
doi = {10.1002/admi.201600781},
year = {2017},
date = {2017-01-23},
journal = {Advanced Materials Interfaces},
volume = {4},
pages = {1600781},
abstract = {The liquid inside a solid material is one of the most common composite materials in nature. The interface between solid–liquid plays an important role in unique deformation. Here, model systems of two polymers (polydimethylsiloxane–polyvinylidenefluoride) are used to make sphere of solid with liquid inside it.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Oliveira, Eliezer Fernando; Pedro Alves da Silva Autreto,; Galvao, Douglas Soares
Silver Hardening via Hypersonic Impacts Online
2017, (preprint arXiv:1801.04780).
@online{Oliveira2017,
title = {Silver Hardening via Hypersonic Impacts},
author = {Eliezer Fernando Oliveira and Pedro Alves da Silva Autreto, and Douglas Soares Galvao},
url = {https://arxiv.org/abs/1801.04780},
year = {2017},
date = {2017-01-15},
abstract = {The search for new ultra strong materials has been a very active research area. With relation
to metals, a successful way to improve their strength is by the creation of a gradient of
nanograins (GNG) inside the material. Recently, R. Thevamaran et al. [Science v354, 312-
316 (2016)] propose a single step method based on high velocity impact of silver nanocubes
to produce high-quality GNG. This method consists of producing high impact collisions of
silver cubes at hypersonic velocity (~400 m/s) against a rigid wall. Although they observed an
improvement in the mechanical properties of the silver after the impact, the GNG creation
and the strengthening mechanism at nanoscale remain unclear. In order to gain further
insights about these mechanisms, we carried out fully atomistic molecular dynamics
simulations (MD) to investigate the atomic conformations/rearrangements during and after
high impact collisions of silver nanocubes at ultrasonic velocity. Our results indicate the coexistence
of polycrystalline arrangements after the impact formed by core HCP domains
surrounded by FCC ones, which could also contribute to explain the structural hardening.},
note = {preprint arXiv:1801.04780},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
to metals, a successful way to improve their strength is by the creation of a gradient of
nanograins (GNG) inside the material. Recently, R. Thevamaran et al. [Science v354, 312-
316 (2016)] propose a single step method based on high velocity impact of silver nanocubes
to produce high-quality GNG. This method consists of producing high impact collisions of
silver cubes at hypersonic velocity (~400 m/s) against a rigid wall. Although they observed an
improvement in the mechanical properties of the silver after the impact, the GNG creation
and the strengthening mechanism at nanoscale remain unclear. In order to gain further
insights about these mechanisms, we carried out fully atomistic molecular dynamics
simulations (MD) to investigate the atomic conformations/rearrangements during and after
high impact collisions of silver nanocubes at ultrasonic velocity. Our results indicate the coexistence
of polycrystalline arrangements after the impact formed by core HCP domains
surrounded by FCC ones, which could also contribute to explain the structural hardening.
Alves, Ana Paula P; Koizumi, Ryota; Samanta, Atanu; Machado, Leonardo D; Singh, Abhisek K; Galvao, Douglas S; Silva, Glaura G; Tiwary, Chandra S; Ajayan, Pulickel M
One-step electrodeposited 3D-ternary composite of zirconia nanoparticles, rGO and polypyrrole with enhanced supercapacitor performance Journal Article
In: Nano Energy, vol. 31, pp. 225-232, 2017.
@article{Alves2017,
title = {One-step electrodeposited 3D-ternary composite of zirconia nanoparticles, rGO and polypyrrole with enhanced supercapacitor performance},
author = {Alves, Ana Paula P and Koizumi, Ryota and Samanta, Atanu and Machado, Leonardo D and Singh, Abhisek K and Galvao, Douglas S and Silva, Glaura G and Tiwary, Chandra S and Ajayan, Pulickel M},
url = {http://www.sciencedirect.com/science/article/pii/S221128551630502X},
doi = {10.1016/j.nanoen.2016.11.018},
year = {2017},
date = {2017-01-01},
journal = {Nano Energy},
volume = {31},
pages = {225-232},
abstract = {Supercapacitor electrodes consisting of conjugated polymers (CP), metal oxides and graphene nanosheets have been explored as a strategy to achieve high specific capacitance, power, energy density, and stability. In this work, we synthesized a 3D structure composed of zirconia oxide nanoparticles (ZrO2), reduced graphene oxide (rGO) and polypyrrole (PPy), using a simple and easily scalable one-step chronopotentiometry method. Detailed characterization revealed that the addition of rGO and ZrO2 modified the morphology of the electrode material. The capacitance of the resulting architecture improved by up to a 100%. The ternary composite featured high stability, with an increase of 5% in capacitance after a thousand cycles. DFT and MD simulations were carried out in order to provide further insight on the role of zirconia.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Chandra Sekhar Tiwary Sujin P Jose, Suppanat Kosolwattana
Enhanced supercapacitor performance of a 3D architecture tailored using atomically thin rGO–MoS 2 2D sheets Journal Article
In: RSC Advances, vol. 6, pp. 93384-93393, 2016.
@article{Jose2016,
title = {Enhanced supercapacitor performance of a 3D architecture tailored using atomically thin rGO–MoS 2 2D sheets},
author = {Sujin P Jose, Chandra Sekhar Tiwary, Suppanat Kosolwattana, Prasanth Raghavan, Leonardo D Machado, Chandkiram Gautam, T Prasankumar, Jarin Joyner, Sehmus Ozden, Douglas S Galvao, PM Ajayan},
url = {xlink.rsc.org/?DOI=c6ra20960b},
doi = {10.1039/C6RA20960B},
year = {2016},
date = {2016-09-19},
journal = {RSC Advances},
volume = {6},
pages = {93384-93393},
abstract = {A 3D architecture is fabricated using 2D nano-sheets of GO and MoS2 as the building blocks by a facile, one-pot chronoamperometry method to achieve a conductive additive free, binder free and scalable supercapacitor electrode. The superior electrochemical properties of the 3D PPy-rGO–MoS2 (PGMo) are due to its porous structure, thin wall, high surface area and high electrical conductivity that endow rapid transportation of electrolyte ions and electrons throughout the electrode matrix. The synergistic effect between the components in a proper ratio improves the supercapacitor performance and material stability of PGMo. The possible correlation of the structure and electrochemical performance of the 3D ternary composite is backed by a fully atomistic molecular dynamics (MD) simulation study. The high specific capacitance (387 F g−1) and impressive cycling stability (>1000 cycles) estimated for PGMo open up an opportunity to consider the 3D ternary nanostructures as cutting edge materials for energy storage solutions.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
P. M. Gautam, Chandkiram; Tiwary, Chandra Sekhar; Machado, Leonardo D.; Jose, Sujin; Ozden, Sehmus; Biradar, Santoshkumar; Galvao, Douglas S.; Sonker, Rakesh K.; Yadav, B. C.; Vajtai, Robert; Ajayan,
Synthesis and porous h-BN 3D architectures for effective humidity and gas sensors Authors Journal Article
In: RSC Advances, vol. 6, no. 91, pp. 87888-87896, 2016.
@article{Gautam2016,
title = {Synthesis and porous h-BN 3D architectures for effective humidity and gas sensors Authors},
author = {P. M. Gautam, Chandkiram and Tiwary, Chandra Sekhar and Machado, Leonardo D. and Jose, Sujin and Ozden, Sehmus and Biradar, Santoshkumar and Galvao, Douglas S. and Sonker, Rakesh K. and Yadav, B. C. and Vajtai, Robert and Ajayan},
url = {pubs.rsc.org/en/Content/ArticleHtml/2016/RA/c6ra18833h},
doi = {10.1039/C6RA18833H},
year = {2016},
date = {2016-09-09},
journal = {RSC Advances},
volume = {6},
number = {91},
pages = {87888-87896},
abstract = {3D (three dimensional) architectures synthesised using an easily scalable solid state method which results in an interconnected network of porous h-BN sheets with boron trioxide are reported in this study. The boron trioxide acts as a nucleating agent for the formation of laterally large nanosheets of h-BN with a low density and increases the specific surface area. The stable form shows improved mechanical properties (experimentally and using MD simulation) and serves as a suitable material for humidity and liquefied petroleum gas (LPG) sensor applications. The sensor shows stability for up to several months without losing its sensitivity.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Leonardo D Machado Sehmus Ozden, ChandraSekhar Tiwary
Ballistic Fracturing of Carbon Nanotubes Journal Article
In: ACS Applied Materials & Interfaces, vol. 8, no. 37, pp. 24819-24825, 2016.
@article{Ozden2016b,
title = {Ballistic Fracturing of Carbon Nanotubes},
author = {Sehmus Ozden, Leonardo D Machado, ChandraSekhar Tiwary, Pedro AS Autreto, Robert Vajtai, Enrique V Barrera, Douglas S Galvao, Pulickel M Ajayan},
url = {pubs.acs.org/doi/abs/10.1021/acsami.6b07547},
doi = {10.1021/acsami.6b07547},
year = {2016},
date = {2016-09-08},
journal = {ACS Applied Materials & Interfaces},
volume = {8},
number = {37},
pages = {24819-24825},
abstract = {Advanced materials with multifunctional capabilities and high resistance to hypervelocity impact are of great interest to the designers of aerospace structures. Carbon nanotubes (CNTs) with their lightweight and high strength properties are alternative to metals and/or metallic alloys conventionally used in aerospace applications. Here we report a detailed study on the ballistic fracturing of CNTs for different velocity ranges. Our results show that the highly energetic impacts cause bond breakage and carbon atom rehybridizations, and sometimes extensive structural reconstructions were also observed. Experimental observations show the formation of nanoribbons, nanodiamonds, and covalently interconnected nanostructures, depending on impact conditions. Fully atomistic reactive molecular dynamics simulations were also carried out in order to gain further insights into the mechanism behind the transformation of CNTs. The simulations show that the velocity and relative orientation of the multiple colliding nanotubes are critical to determine the impact outcome.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Chandra Sekhar Tiwary Mohamad A Kabbani, Anirban Som
A generic approach for mechano-chemical reactions between carbon nanotubes of different functionalities Journal Article
In: Carbon, vol. 104, pp. 196-202, 2016.
@article{Kabbani2016,
title = {A generic approach for mechano-chemical reactions between carbon nanotubes of different functionalities},
author = {Mohamad A Kabbani, Chandra Sekhar Tiwary, Anirban Som, KR Krishnadas, Pedro AS Autreto, Sehmus Ozden, Kunttal Keyshar, Ken Hackenberg, Alin Christian Chipara, Douglas S Galvao, Robert Vajtai, Ahmad T Kabbani, Thalappil Pradeep, Pulickel M Ajayan},
url = {www.sciencedirect.com/science/article/pii/S000862231630183X},
doi = {10.1016/j.carbon.2016.02.094},
year = {2016},
date = {2016-08-31},
journal = {Carbon},
volume = {104},
pages = {196-202},
abstract = {Abstract Here, we report similar reactions between nanotubes carrying functionalities,
namely carbon nanotubes (CNTs) with the acyl chloride/hydroxyl and amine/carboxylic
functionalities directly attached to their surfaces, resulting in the formation ofchemically
modified graphene products. The reaction is spontaneous and is facilitated by simple
grinding of the reactants. The new solid-state reactions have been confirmed using different
spectroscopic and electron microscopy techniques.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
namely carbon nanotubes (CNTs) with the acyl chloride/hydroxyl and amine/carboxylic
functionalities directly attached to their surfaces, resulting in the formation ofchemically
modified graphene products. The reaction is spontaneous and is facilitated by simple
grinding of the reactants. The new solid-state reactions have been confirmed using different
spectroscopic and electron microscopy techniques.
Chandra Sekhar Tiwary Dibyendu Chakravarty, Cristano F Woellner
3D Porous Graphene by Low-Temperature Plasma Welding for Bone Implants Journal Article
In: Advanced Materials, vol. 28, no. 40, pp. 8959-8967, 2016.
@article{chakravarty20163d,
title = {3D Porous Graphene by Low-Temperature Plasma Welding for Bone Implants},
author = {Dibyendu Chakravarty, Chandra Sekhar Tiwary, Cristano F Woellner, Sruthi Radhakrishnan, Soumya Vinod, Sehmus Ozden, Pedro Alves da Silva Autreto, Sanjit Bhowmick, Syed Asif, Sendurai A Mani, Douglas S Galvao, Pulickel M},
url = {onlinelibrary.wiley.com/doi/10.1002/adma.201603146/abstract },
doi = {10.1002/adma.201603146},
year = {2016},
date = {2016-08-26},
journal = {Advanced Materials},
volume = {28},
number = {40},
pages = {8959-8967},
abstract = {3D scaffolds of graphene, possessing ultra-low density, macroporous microstructure, and high yield strength and stiffness can be developed by a novel plasma welding process. The bonding between adjacent graphene sheets is investigated by molecular dynamics simulations. The high degree of biocompatibility along with high porosity and good mechanical properties makes graphene an ideal material for use as body implants.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Amelia HC Hart Ryota Koizumi, Gustavo Brunetto
Mechano-chemical stabilization of three-dimensional carbon nanotube aggregates Journal Article
In: Carbon, vol. 110, pp. 27-33, 2016.
@article{koizumi2016mechano,
title = {Mechano-chemical stabilization of three-dimensional carbon nanotube aggregates},
author = {Ryota Koizumi, Amelia HC Hart, Gustavo Brunetto, Sanjit Bhowmick, Peter S Owuor, John T Hamel, Anieph X Gentles, Sehmus Ozden, Jun Lou, Robert Vajtai, SA Syed Asif, Douglas S Galvão, CS Tiwary, PM Ajayan},
url = {www.sciencedirect.com/science/article/pii/S0008622316307400},
doi = {10.1016/j.carbon.2016.08.085},
year = {2016},
date = {2016-08-21},
journal = {Carbon},
volume = {110},
pages = {27-33},
abstract = {Here we report a combined study of experiments and simulations to understand how chemical functional groups can mechanically stabilize aggregates of carbon nanotubes (CNTs). Ultralow density aggregates of chemically functionalized CNTs, in the form of macro-scale spheres made by freeze-drying method, show mechanical stabilization and near complete elastic recovery during deformation. Simulations of interacting functionalized carbon nanotube aggregates show better structural retention compared to non-functionalized CNTs under compression, suggesting that the atomic-level interactions between functional groups on adjoining CNTs help maintain structural rigidity and elastic response during loading. Aggregates of non-functionalized CNTs collapses under similar loading conditions. The dynamic mechanical responses of CNT macrostructures and mechano-chemical stabilization are directly observed using in-situ deformation inside a scanning electron microscope.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Chandra Sekhar Tiwary Soumya Vinod, Leonardo D Machado
Synthesis of ultralow density 3D graphene–CNT foams using a two-step method Journal Article
In: Nanoscale, vol. 8, no. 35, pp. 15857-15863, 2016.
@article{Vinod2016b,
title = {Synthesis of ultralow density 3D graphene–CNT foams using a two-step method},
author = {Soumya Vinod, Chandra Sekhar Tiwary, Leonardo D Machado, Sehmus Ozden, Robert Vajtai, Douglas S Galvao, Pulickel M Ajayan},
url = {xlink.rsc.org/?DOI=c6nr04252j},
doi = {10.1039/C6NR04252J},
year = {2016},
date = {2016-08-09},
journal = {Nanoscale},
volume = {8},
number = {35},
pages = {15857-15863},
abstract = {Here, we report a highly scalable two-step method to produce graphene foams with ordered carbon nanotube reinforcements. In our approach, we first used solution assembly methods to obtain graphene oxide foam. Next, we employed chemical vapor deposition to simultaneously grow carbon nanotubes and thermally reduce the 3D graphene oxide scaffold. The resulting structure presented increased stiffness, good mechanical stability and oil absorption properties. Molecular dynamics simulations were carried out to further elucidate failure mechanisms and to understand the enhancement of the mechanical properties. The simulations showed that mechanical failure is directly associated with bending of vertical reinforcements, and that, for similar length and contact area, much more stress is required to bend the corresponding reinforcements of carbon nanotubes, thus explaining the experimentally observed enhanced mechanical properties.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
T Botari JM de Sousa, E Perim
Mechanical and structural properties of graphene-like carbon nitride sheets Journal Article
In: RSC Advances, vol. 6, no. 80, pp. 76915-76921, 2016.
@article{deSousa2016b,
title = {Mechanical and structural properties of graphene-like carbon nitride sheets},
author = {JM de Sousa, T Botari, E Perim, RA Bizao, Douglas S Galvao},
url = {pubs.rsc.org/en/content/articlehtml/2016/ra/c6ra14273g},
doi = {10.1039/C6RA14273G},
year = {2016},
date = {2016-08-08},
journal = {RSC Advances},
volume = {6},
number = {80},
pages = {76915-76921},
abstract = {Carbon nitride-based nanostructures have attracted special attention (from theory and experiments) due to their remarkable electromechanical properties. In this work we have investigated the mechanical properties of some graphene-like carbon nitride membranes through fully atomistic reactive molecular dynamics simulations. We have analyzed three different structures of these CN families, the so-called graphene-based g-CN, triazine-based g-C3N4 and heptazine-based g-C3N4. The stretching dynamics of these membranes was studied for deformations along their two main axes and at three different temperatures: 10 K, 300 K and 600 K. We show that g-CN membranes have the lowest ultimate fracture strain value, followed by heptazine-based and triazine-based ones, respectively. This behavior can be explained in terms of their differences in density values, topologies and types of chemical bonds. The dependency of the fracture patterns on the stretching directions is also discussed.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Rodrigo Prioli Clara M Almeida, Benjamin Fragneaud
Giant and Tunable Anisotropy of Nanoscale Friction in Graphene Journal Article
In: Nature Scientific Reports, vol. 6, pp. 31569, 2016.
@article{Almeida2016,
title = {Giant and Tunable Anisotropy of Nanoscale Friction in Graphene},
author = {Clara M Almeida, Rodrigo Prioli, Benjamin Fragneaud, Luiz Gustavo Cançado, Ricardo Paupitz, Douglas S Galvão, Marcelo De Cicco, Marcos G Menezes, Carlos A Achete, Rodrigo B Capaz},
url = {http://www-nature-com.ez88.periodicos.capes.gov.br/articles/srep31569},
doi = {10.1038/srep31569},
year = {2016},
date = {2016-07-18},
journal = {Nature Scientific Reports},
volume = {6},
pages = {31569},
abstract = {The nanoscale friction between an atomic force microscopy tip and graphene is investigated using friction force microscopy (FFM). During the tip movement, friction forces are observed to increase and then saturate in a highly anisotropic manner. As a result, the friction forces in graphene are highly dependent on the scanning direction: under some conditions, the energy dissipated along the armchair direction can be 80% higher than along the zigzag direction. In comparison, for highly-oriented pyrolitic graphite (HOPG), the friction anisotropy between armchair and zigzag directions is only 15%. This giant friction anisotropy in graphene results from anisotropies in the amplitudes of flexural deformations of the graphene sheet driven by the tip movement, not present in HOPG. The effect can be seen as a novel manifestation of the classical phenomenon of Euler buckling at the nanoscale, which provides the non-linear ingredients that amplify friction anisotropy. Simulations based on a novel version of the 2D Tomlinson model (modified to include the effects of flexural deformations), as well as fully atomistic molecular dynamics simulations and first-principles density-functional theory (DFT) calculations, are able to reproduce and explain the experimental observations.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Pedro Alves da Silva Autreto Cristiano Francisco Woellner, Douglas S Galvao
Graphone (one-side hydrogenated graphene) formation on different substrates Online
2016.
@online{Woellner2016b,
title = {Graphone (one-side hydrogenated graphene) formation on different substrates},
author = {Cristiano Francisco Woellner, Pedro Alves da Silva Autreto, Douglas S Galvao},
url = {arXiv preprint arXiv:1606.09235},
year = {2016},
date = {2016-06-29},
abstract = {In this work we present a fully atomistic reactive (ReaxFF force field) molecular dynamics study of the structural and dynamical aspects of the one-side hydrogenation of graphene membranes, leading to the formation of the so-called graphone structure. We have considered different substrates: graphene, few-layers graphene, graphite and platinum at different temperatures. Our results showed that the hydrogenation rates are very dependent on the substrate and thermal effects. Our results also showed that, similarly to graphane, large hydrogenated domains are unlikely to be formed. These hydrogenation processes occur through the formation of uncorrelated cluster domains.},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Sehmus Ozden Leonardo D Machado, ChandraSekhar Tiwary
The structural and dynamical aspects of boron nitride nanotubes under high velocity impacts Journal Article
In: Physical Chemistry Chemical Physics, vol. 18, pp. 14776-14781, 2016.
@article{Machado2016,
title = {The structural and dynamical aspects of boron nitride nanotubes under high velocity impacts},
author = {Leonardo D Machado, Sehmus Ozden, ChandraSekhar Tiwary, Pedro AS Autreto, Robert Vajtai, Enrique V Barrera, Douglas S Galvao, Pulickel M Ajayan},
url = {xlink.rsc.org/?DOI=c6cp01949h},
doi = {10.1039/C6CP01949H},
year = {2016},
date = {2016-05-01},
journal = {Physical Chemistry Chemical Physics},
volume = {18},
pages = {14776-14781},
abstract = {This communication report is a study on the structural and dynamical aspects of boron nitride nanotubes (BNNTs) shot at high velocities (∼5 km s−1) against solid targets. The experimental results show unzipping of BNNTs and the formation of hBN nanoribbons. Fully atomistic reactive molecular dynamics simulations were also carried out to gain insights into the BNNT fracture patterns and deformation mechanisms. Our results show that longitudinal and axial tube fractures occur, but the formation of BN nanoribbons from fractured tubes was only observed for some impact angles. Although some structural and dynamical features of the impacts are similar to the ones reported for CNTs, because BNNTs are more brittle than CNTs this results in a larger number of fractured tubes but with fewer formed nanoribbons.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Botari, Tiago; Paupitz, Ricardo; da Silva Autreto, Pedro Alves; Galvao, Douglas S
Graphene healing mechanisms: A theoretical investigation Journal Article
In: Carbon, vol. 99, pp. 302-309, 2016.
@article{2016Healing,
title = {Graphene healing mechanisms: A theoretical investigation},
author = {Botari, Tiago and Paupitz, Ricardo and da Silva Autreto, Pedro Alves and Galvao, Douglas S},
url = {http://www.sciencedirect.com/science/article/pii/S0008622315304784},
doi = {10.1016/j.carbon.2015.11.070},
year = {2016},
date = {2016-04-30},
journal = {Carbon},
volume = {99},
pages = {302-309},
abstract = {Large holes in graphene membranes were recently shown to heal, either at room temperature during a low energy STEM experiment, or by annealing at high temperatures. However, the details of the healing mechanism remain unclear. We carried out fully atomistic reactive molecular dynamics simulations in order to address these mechanisms under different experimental conditions. Our results show that, if a carbon atom source is present, high temperatures can provide enough energy for the carbon atoms to overcome the potential energy barrier and to produce perfect reconstruction of the graphene hexagonal structure. At room temperature, this perfect healing is only possible if the heat effects of the electron beam from STEM experiment are explicitly taken into account. The reconstruction process of a perfect or near perfect graphene structure involves the formation of linear carbon chains, as well as rings containing 5, 6, 7 and 8 atoms with planar (Stone-Wales like) and non-planar (lump like) structures. These results shed light on the healing mechanism of graphene when subjected to different experimental conditions. Additionally, the methodology presented here can be useful for investigating the tailoring and manipulations of other nano-structures.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2005
Legoas, SB; Rodrigues, V; Ugarte, D; Galvao, DS
Legoas et al. Reply Journal Article
In: Physical Review Letters, vol. 95, no. 16, pp. 169602, 2005.
Links | BibTeX | Tags: Linear Atomic Chains, Metallic Nanowires, Molecular Dynamics
@article{legoas2005legoas,
title = {Legoas et al. Reply},
author = {Legoas, SB and Rodrigues, V and Ugarte, D and Galvao, DS},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.95.169602},
year = {2005},
date = {2005-01-01},
journal = {Physical Review Letters},
volume = {95},
number = {16},
pages = {169602},
keywords = {Linear Atomic Chains, Metallic Nanowires, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}

Coluci, Vitor; Braga, Scheila F; Baughman, Ray H; Galvao, Douglas S
Hydrogen Storage in Carbon Nanoscrolls: A Molecular Dynamics Study Proceedings
Cambridge University Press, vol. 885, 2005.
Abstract | Links | BibTeX | Tags: Hydrogen Storage, Molecular Dynamics, Monte Carlo, Scrolls
@proceedings{coluci2005hydrogen,
title = {Hydrogen Storage in Carbon Nanoscrolls: A Molecular Dynamics Study},
author = {Coluci, Vitor and Braga, Scheila F and Baughman, Ray H and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8012272&fileId=S1946427400039816},
year = {2005},
date = {2005-01-01},
journal = {MRS Proceedings},
volume = {885},
pages = {0885--A06},
publisher = {Cambridge University Press},
abstract = {We carried out molecular dynamics simulations with Tersoff-Brenner potentials in order to investigate the hydrogen uptake mechanisms and storage capacity of carbon nanoscrolls (CNSs). CNSs are jelly roll-like structures formed by wrapping graphene layers. Interlayer adsorption is an option for this material, which does not exist for single and multiwalled carbon nanotubes. We analyzed the processes of hydrogen physisorption and uptake mechanisms. We observed incorporation of hydrogen molecules in both external and internal scroll surfaces. Insertion in the internal cavity and between the scroll layers is responsible for 40% of the total hydrogen adsorption at 77 K.},
keywords = {Hydrogen Storage, Molecular Dynamics, Monte Carlo, Scrolls},
pubstate = {published},
tppubtype = {proceedings}
}
2004

Braga, Scheila F; Coluci, Vitor R; Legoas, Sergio B; Giro, Ronaldo; Galvao, Douglas S; Baughman, Ray H
Structure and dynamics of carbon nanoscrolls Journal Article
In: Nano Letters, vol. 4, no. 5, pp. 881–884, 2004.
Abstract | Links | BibTeX | Tags: Molecular Dynamics, Scrolls, Structure
@article{braga2004structure,
title = {Structure and dynamics of carbon nanoscrolls},
author = {Braga, Scheila F and Coluci, Vitor R and Legoas, Sergio B and Giro, Ronaldo and Galvao, Douglas S and Baughman, Ray H},
url = {http://pubs.acs.org/doi/abs/10.1021/nl0497272},
year = {2004},
date = {2004-01-01},
journal = {Nano Letters},
volume = {4},
number = {5},
pages = {881--884},
publisher = {American Chemical Society},
abstract = {Carbon nanotube scrolls (CNSs) provide an interesting form of carbon that ideally consists of a single sheet of graphite that is spiral wrapped
to form a nanotube. We here use molecular dynamics simulations to investigate CNS formation, stability, and the structural effects due to
charge injection. CNS formation is seen to automatically occur when a critical overlap between sheet layers is achieved for the partially curled
sheet. We find that charge injection causes unwinding of the CNSs, which might be important for the application of CNSs as nanomechanical
actuators},
keywords = {Molecular Dynamics, Scrolls, Structure},
pubstate = {published},
tppubtype = {article}
}
to form a nanotube. We here use molecular dynamics simulations to investigate CNS formation, stability, and the structural effects due to
charge injection. CNS formation is seen to automatically occur when a critical overlap between sheet layers is achieved for the partially curled
sheet. We find that charge injection causes unwinding of the CNSs, which might be important for the application of CNSs as nanomechanical
actuators

Legoas, SB; Coluci, VR; Braga, SF; Coura, PZ; Dantas, SO; Galvao, DS
Gigahertz nanomechanical oscillators based on carbon nanotubes Journal Article
In: Nanotechnology, vol. 15, no. 4, pp. S184, 2004.
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, Molecular Dynamics, Oscillators
@article{legoas2004gigahertz,
title = {Gigahertz nanomechanical oscillators based on carbon nanotubes},
author = {Legoas, SB and Coluci, VR and Braga, SF and Coura, PZ and Dantas, SO and Galvao, DS},
url = {http://iopscience.iop.org/0957-4484/15/4/012},
year = {2004},
date = {2004-01-01},
journal = {Nanotechnology},
volume = {15},
number = {4},
pages = {S184},
publisher = {IOP Publishing},
abstract = {We report molecular dynamics studies of carbon nanotubes as mechanical gigahertz oscillators. Our results show that different oscillatory regimes exist but that sustained oscillations are possible only when the radii difference values of the inner and outer tubes are {sim }3.4~AA . Frequencies as large as 87 GHz were obtained. Calculated force and frequency values are in good agreement with estimated data from recent experimental investigations.},
keywords = {Carbon Nanotubes, Molecular Dynamics, Oscillators},
pubstate = {published},
tppubtype = {article}
}
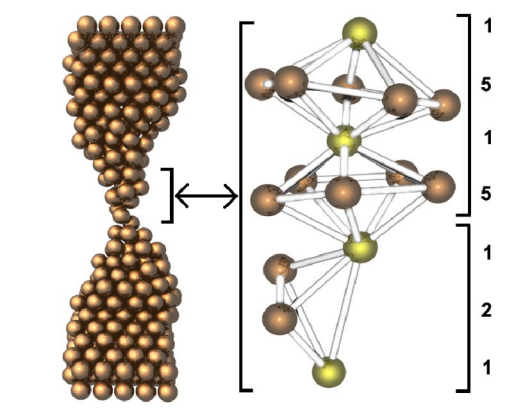
Gonzalez, JC; Rodrigues, V; Bettini, J; Rego, LGC; Rocha, AR; Coura, PZ; Dantas, SO; Sato, F; Galvao, DS; Ugarte, D
Indication of unusual pentagonal structures in atomic-size Cu nanowires Journal Article
In: Physical Review Letters, vol. 93, no. 12, pp. 126103, 2004.
Abstract | Links | BibTeX | Tags: Copper Nanowires, Linear Atomic Chains, Metallic Nanowires, Molecular Dynamics, TEM, top20
@article{gonzalez2004indication,
title = {Indication of unusual pentagonal structures in atomic-size Cu nanowires},
author = {Gonzalez, JC and Rodrigues, V and Bettini, J and Rego, LGC and Rocha, AR and Coura, PZ and Dantas, SO and Sato, F and Galvao, DS and Ugarte, D},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.93.126103},
year = {2004},
date = {2004-01-01},
journal = {Physical Review Letters},
volume = {93},
number = {12},
pages = {126103},
publisher = {APS},
abstract = {We present a study of the structural and quantum conductance properties of atomic-size copper nanowires generated by mechanical stretching. The atomistic evolution was derived from time-resolved electron microscopy observations and molecular dynamics simulations. We have analyzed the quantum transport behavior by means of conductance measurements and theoretical calculations. The results suggest the formation of an unusual and highly stable pentagonal Cu nanowire with a diameter of ∼0.45 nm and ∼4.5 conductance quanta.
},
keywords = {Copper Nanowires, Linear Atomic Chains, Metallic Nanowires, Molecular Dynamics, TEM, top20},
pubstate = {published},
tppubtype = {article}
}
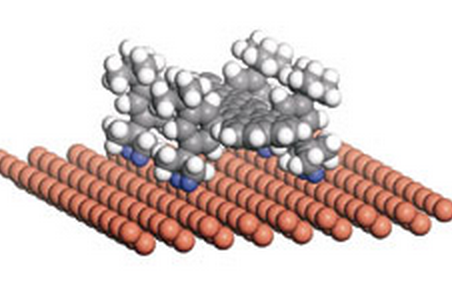
Otero, Roberto; Hummelink, Frauke; Sato, Fernando; Legoas, Sergio B; Thostrup, Peter; Lægsgaard, Erik; Stensgaard, Ivan; Galvao, Douglas S; Besenbacher, Flemming
Lock-and-key effect in the surface diffusion of large organic molecules probed by STM Journal Article
In: Nature Materials, vol. 3, no. 11, pp. 779–782, 2004.
Abstract | Links | BibTeX | Tags: Landers, Molecular Dynamics, Molecular Electronics, STM, top20
@article{otero2004lock,
title = {Lock-and-key effect in the surface diffusion of large organic molecules probed by STM},
author = {Otero, Roberto and Hummelink, Frauke and Sato, Fernando and Legoas, Sergio B and Thostrup, Peter and Lægsgaard, Erik and Stensgaard, Ivan and Galvao, Douglas S and Besenbacher, Flemming},
url = {http://www.nature.com/nmat/journal/v3/n11/full/nmat1243.html},
year = {2004},
date = {2004-01-01},
journal = {Nature Materials},
volume = {3},
number = {11},
pages = {779--782},
publisher = {Nature Publishing Group},
abstract = {A nanoscale understanding of the complex dynamics of large molecules at surfaces is essential for the bottom-up design of molecular nanostructures1, 2, 3, 4, 5, 6, 7, 8. Here we show that we can change the diffusion coefficient of the complex organic molecule known as Violet Lander (VL, C108H104) on Cu(110) by two orders of magnitude by using the STM at low temperatures to switch between two adsorption configurations that differ only in the molecular orientation with respect to the substrate lattice. From an interplay with molecular dynamics simulations, we interpret the results within a lock-and-key model similar to the one driving the recognition between biomolecules: the molecule (key) is immobilized only when its orientation is such that the molecular shape fits the atomic lattice of the surface (lock); otherwise the molecule is highly mobile.
Introduction
},
keywords = {Landers, Molecular Dynamics, Molecular Electronics, STM, top20},
pubstate = {published},
tppubtype = {article}
}
Introduction
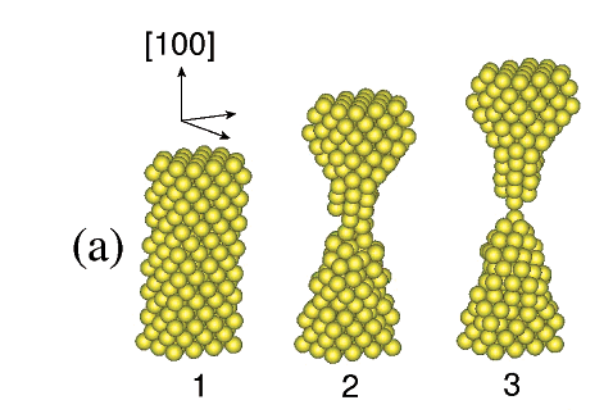
Coura, Pablo Z; Legoas, Sergio B; Moreira, Anderson S; Sato, Fernando; Rodrigues, Varlei; Dantas, Socrates O; Ugarte, Daniel; Galvao, Douglas S
On the structural and stability features of linear atomic suspended chains formed from gold nanowires stretching Journal Article
In: Nano Letters, vol. 4, no. 7, pp. 1187–1191, 2004.
Abstract | Links | BibTeX | Tags: Liinear Atomic Chains, Metallic Nanowires, Molecular Dynamics, Structure, TEM
@article{coura2004structural,
title = {On the structural and stability features of linear atomic suspended chains formed from gold nanowires stretching},
author = {Coura, Pablo Z and Legoas, Sergio B and Moreira, Anderson S and Sato, Fernando and Rodrigues, Varlei and Dantas, Socrates O and Ugarte, Daniel and Galvao, Douglas S},
url = {http://pubs.acs.org/doi/abs/10.1021/nl049725h},
year = {2004},
date = {2004-01-01},
journal = {Nano Letters},
volume = {4},
number = {7},
pages = {1187--1191},
publisher = {American Chemical Society},
abstract = {Metallic nanowires (NWs) have been intensily investigated in the past years, but details on
their formation are still not completely understood. In this work we report high resolution
transmission electron microscopy data and molecular dynamics simulation results for gold
NW elongation. Our results show that different initial crystallographic orientations lead to
very differentiated linear atomic suspended chain (LAC) formations and strongly support that
kinetic aspects are the dominant mechanisms determining the LAC morphologies.},
keywords = {Liinear Atomic Chains, Metallic Nanowires, Molecular Dynamics, Structure, TEM},
pubstate = {published},
tppubtype = {article}
}
their formation are still not completely understood. In this work we report high resolution
transmission electron microscopy data and molecular dynamics simulation results for gold
NW elongation. Our results show that different initial crystallographic orientations lead to
very differentiated linear atomic suspended chain (LAC) formations and strongly support that
kinetic aspects are the dominant mechanisms determining the LAC morphologies.
Legoas, Sergio B; Giro, Ronaldo; Galvao, Douglas S
Molecular dynamics simulations of C6) nanobearings Journal Article
In: Chemical physics letters, vol. 386, no. 4, pp. 425–429, 2004.
Abstract | Links | BibTeX | Tags: C60, Fullerenes, Molecular Dynamics, Nanobearing, Tribology
@article{legoas2004molecular,
title = {Molecular dynamics simulations of C6) nanobearings},
author = {Legoas, Sergio B and Giro, Ronaldo and Galvao, Douglas S},
url = {http://www.sciencedirect.com/science/article/pii/S000926140400168X},
year = {2004},
date = {2004-01-01},
journal = {Chemical physics letters},
volume = {386},
number = {4},
pages = {425--429},
publisher = {Elsevier},
abstract = {Recently was reported an ultra-lubricated system based on C60 molecules deposited over graphite layers. In that work a stick-slip rolling model for C60 molecules was proposed to explain the observed ultra-low friction force. In this Letter, we report the first molecular dynamics studies for these systems. Our results show that the AB stacking is not observed and the main experimental features can be explained without invoking stick-slip motions.
},
keywords = {C60, Fullerenes, Molecular Dynamics, Nanobearing, Tribology},
pubstate = {published},
tppubtype = {article}
}
2003

VR Coluci SB Legoas, SF Braga
Molecular-dynamics simulations of carbon nanotubes as gigahertz oscillators Journal Article
In: Physical Review Letters, vol. 90, no. 5, pp. 055504, 2003.
Abstract | BibTeX | Tags: Carbon Nanotubes, Molecular Dynamics, Oscillators, top20
@article{legoas2003molecularb,
title = {Molecular-dynamics simulations of carbon nanotubes as gigahertz oscillators},
author = {SB Legoas, VR Coluci, SF Braga, PZ Coura, SO Dantas, DS Galvao},
year = {2003},
date = {2003-01-01},
journal = {Physical Review Letters},
volume = {90},
number = {5},
pages = {055504},
abstract = {Recently, Zheng and Jiang [Phys. Rev. Lett. 88, 045503 (2002)] have proposed that multiwalled carbon nanotubes could be the basis for a new generation of nano-oscillators in the several gigahertz range. In this Letter, we present the first molecular dynamics simulation for these systems. Different nanotube types were considered in order to verify the reliability of such devices as gigahertz oscillators. Our results show that these nano-oscillators are dynamically stable when the radii difference values between inner and outer tubes are of ∼3.4 Å. Frequencies as large as 38 GHz were observed, and the calculated force values are in good agreement with recent experimental investigations.},
keywords = {Carbon Nanotubes, Molecular Dynamics, Oscillators, top20},
pubstate = {published},
tppubtype = {article}
}
1993
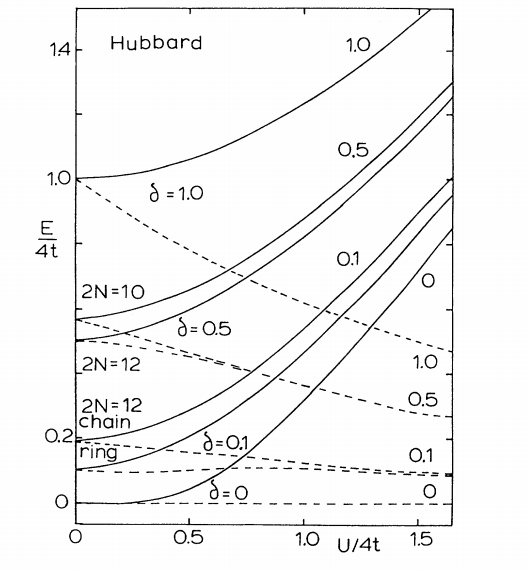
S Ramasesha ZG Soos, DS Galvao
In: Physical Review Letters, vol. 71, no. 10, pp. 1609, 1993.
Abstract | Links | BibTeX | Tags: Molecular Dynamics
@article{soos1993band,
title = {Band to correlated crossover in alternating Hubbard and Pariser-Parr-Pople chains: Nature of the lowest singlet excitation of conjugated polymers},
author = {ZG Soos, S Ramasesha, DS Galvao},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.71.1609},
doi = {10.1103/PhysRevLett.71.1609},
year = {1993},
date = {1993-01-01},
journal = {Physical Review Letters},
volume = {71},
number = {10},
pages = {1609},
publisher = {American Physical Society},
abstract = {The evolution with increasing Coulomb correlations of a semiconductor to a magnetic insulator is related to an excited-state crossover in π-electron models for conjugated polymers. We associate strong fluorescence with a lowest singlet excitation S1 that is dipole allowed, on the band side, while S1 becomes two-photon allowed on the correlated side. S1/S2 crossovers in Hubbard, Pariser-Parr-Pople, or other chains with electron-hole symmetry and alternating transfer integrals t(1±δ) are based on exact results at δ=0 and 1, on molecular exciton theory at large δ, and on oligomer calculations up to twelve sites.},
keywords = {Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ

