
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ
JM; Sousa, Bizao
Elastic Properties of Graphyne-based Nanotubes Online
2019, (ArXiv preprint.).
@online{deSousa2019b,
title = {Elastic Properties of Graphyne-based Nanotubes},
author = {de Sousa, JM; , Bizao, RA; Sousa Filho, VP; Aguiar, AL; Coluci, VR; Pugno, NM; Girao, EC; Souza Filho, AG; Galvao, DS},
url = {https://arxiv.org/pdf/1905.02104.pdf},
year = {2019},
date = {2019-04-07},
abstract = {Graphyne nanotubes (GNTs) are nanostructures obtained from rolled up graphyne sheets,
in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes
are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to
conventional CNTs, GNTs can present different chiralities and electronic properties. Because
of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their
mechanical properties. In this work, we studied the mechanical response of GNTs under
tensile stress using fully atomistic molecular dynamics simulations and density functional
theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs
at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller
ultimate strength and Young’s modulus values. This is a consequence of the combined
effects of the existence of triple bonds and increased porosity/flexibility due to the presence
of acetylenic groups.},
note = {ArXiv preprint.},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes
are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to
conventional CNTs, GNTs can present different chiralities and electronic properties. Because
of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their
mechanical properties. In this work, we studied the mechanical response of GNTs under
tensile stress using fully atomistic molecular dynamics simulations and density functional
theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs
at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller
ultimate strength and Young’s modulus values. This is a consequence of the combined
effects of the existence of triple bonds and increased porosity/flexibility due to the presence
of acetylenic groups.
JM; Sousa, Bizao
Elastic Properties of Graphyne-Based Nanotubes Journal Article
Em: Computational Materials Science, vol. 170, pp. 109153, 2019.
@article{deSousa2019c,
title = {Elastic Properties of Graphyne-Based Nanotubes},
author = {de Sousa, JM; , Bizao, RA; Sousa Filho, VP; Aguiar, AL; Coluci, VR; Pugno, NM; Girao, EC; Souza Filho, AG; Galvao, DS},
url = {https://www.sciencedirect.com/science/article/pii/S0927025619304525?dgcid=coauthor#s0040},
doi = {10.1016/j.commatsci.2019.109153},
year = {2019},
date = {2019-04-03},
journal = {Computational Materials Science},
volume = {170},
pages = {109153},
abstract = {Graphyne nanotubes (GNTs) are nanostructures obtained from rolled up graphyne sheets, in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to conventional CNTs, GNTs can present different chiralities and electronic properties. Because of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their mechanical properties. In this work, we studied the mechanical response of GNTs under tensile stress using fully atomistic molecular dynamics simulations and density functional theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller ultimate strength and Young’s modulus values. This is a consequence of the combined effects of the existence of triple bonds and increased porosity/flexibility due to the presence of acetylenic groups.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Chipara, A. C.; Tsafack, T.; Owuor, P. S.; Yeon, J.; Junkermeier, C. E.; van Duin, A. C. T.; Bhowmick, S.; Asif, S. A. S.; Radhakrishnan, S.; Park, J. H.; Brunetto, G.; Kaipparettu, B. A.; Galvão, D. S.; Chipara, M.; Lou, J.; Tsang, H. H.; Dubey, M.; Vajtai, R.; Tiwary, C. S.; Ajayan, P. M.
Underwater Adhesive using Solid–liquid Polymer Mixes Journal Article
Em: Materials Today Chemistry, vol. 9, pp. 149-157, 2018.
@article{Chipara2018,
title = {Underwater Adhesive using Solid–liquid Polymer Mixes},
author = {A.C. Chipara and T. Tsafack and P.S. Owuor and J. Yeon and C.E. Junkermeier and A.C.T. van Duin and S. Bhowmick and S.A.S. Asif and S. Radhakrishnan and J.H. Park and G. Brunetto and B.A. Kaipparettu and D.S. Galvão and M. Chipara and J. Lou and H.H. Tsang and M. Dubey and R. Vajtai and C.S. Tiwary and P.M. Ajayan},
url = {https://www.sciencedirect.com/science/article/pii/S2468519418301423#appsec1},
doi = {10.1016/j.mtchem.2018.07.002},
year = {2018},
date = {2018-08-08},
journal = {Materials Today Chemistry},
volume = {9},
pages = {149-157},
abstract = {Instantaneous adhesion between different materials is a requirement for several applications ranging from electronics to biomedicine. Approaches such as surface patterning, chemical cross-linking, surface modification, and chemical synthesis have been adopted to generate temporary adhesion between various materials and surfaces. Because of the lack of curing times, temporary adhesives are instantaneous, a useful property for specific applications that need quick bonding. However, to this day, temporary adhesives have been mainly demonstrated under dry conditions and do not work well in submerged or humid environments. Furthermore, most rely on chemical bonds resulting from strong interactions with the substrate such as acrylate based. This work demonstrates the synthesis of a universal amphibious adhesive solely by combining solid polytetrafluoroethylene (PTFE) and liquid polydimethylsiloxane (PDMS) polymers. While the dipole-dipole interactions are induced by a large electronegativity difference between fluorine atoms in PTFE and hydrogen atoms in PDMS, strong surface wetting allows the proposed adhesive to fully coat both substrates and PTFE particles, thereby maximizing the interfacial chemistry. The two-phase solid–liquid polymer system displays adhesive characteristics applicable both in air and water, and enables joining of a wide range of similar and dissimilar materials (glasses, metals, ceramics, papers, and biomaterials). The adhesive exhibits excellent mechanical properties for the joints between various surfaces as observed in lap shear testing, T-peel testing, and tensile testing. The proposed biocompatible adhesive can also be reused multiple times in different dry and wet environments. Additionally, we have developed a new reactive force field parameterization and used it in our molecular dynamics simulations to validate the adhesive nature of the mixed polymer system with different surfaces. This simple amphibious adhesive could meet the need for a universal glue that performs well with a number of materials for a wide range of conditions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Oliveira, Eliezer F.; Autreto, Pedro A. S.; Woellner, Cristiano F.; Galvao, Douglas S.
On the mechanical properties of novamene: a fully atomistic molecular dynamics and DFT investigation Journal Article
Em: Carbon, vol. 139, pp. 782-788, 2018.
@article{Oliveira2018e,
title = {On the mechanical properties of novamene: a fully atomistic molecular dynamics and DFT investigation},
author = {Eliezer F. Oliveira and Pedro A. S. Autreto and Cristiano F. Woellner and Douglas S. Galvao},
url = {https://www.sciencedirect.com/science/article/pii/S0008622318306882?via%3Dihub#appsec1},
doi = {10.1016/j.carbon.2018.07.038},
year = {2018},
date = {2018-07-19},
journal = {Carbon},
volume = {139},
pages = {782-788},
abstract = {We have investigated through fully atomistic reactive molecular dynamics and density functional theory simulations, the mechanical properties and fracture dynamics of single-ringed novamene (1R-novamene), a new 3D carbon allotrope structure recently proposed. Our results showed that 1R-novamene is an anisotropic structure with relation to tensile deformation. Although 1R-novamente shares some mechanical features with other carbon allotropes, it also exhibits distinct ones, such as, extensive structural reconstructions. 1R-novamene presents ultimate strength (∼100 GPa) values lower than other carbon allotropes, but it has the highest ultimate strain along the z-direction (∼22.5%). Although the Young's modulus (∼600 GPa) and ultimate strength values are smaller than for other carbon allotropes, they still outperform other materials, such as for example silicon, steel or titanium alloys. With relation to the fracture dynamics, 1R-novamene is again anisotropic with the fracture/crack propagation originating from deformed heptagons and pentagons for x and y directions and broken sp3 bonds connecting structural planes. Another interesting feature is the formation of multiple and long carbon linear chains in the final fracture stages.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Balan, Aravind Puthirath; Radhakrishnan, Sruthi; Woellner, Cristiano F.; Sinha, Shyam K.; Deng, Liangzi; de los Reyes, Carlos; Rao, Manmadha; Paulose, Maggie; Neupane, Ram; Vajtai, Robert; Chu, Ching-Wu; Costin, Gelu; Galvao, Douglas S.; Marti, Angel A.; van Aken, Peter; Varghese, Oomman K; Tiwary, Chandra Sekhar; Anantharaman, M R; Ajayan, Pulickel M
Exfoliation of a non-van der Waals material from iron ore hematite Journal Article
Em: Nature Nanotechnology, vol. 13, pp. 602–610, 2018.
@article{Balan2018,
title = {Exfoliation of a non-van der Waals material from iron ore hematite},
author = {Aravind Puthirath Balan and Sruthi Radhakrishnan and Cristiano F. Woellner and Shyam K. Sinha and Liangzi Deng and Carlos de los Reyes and Manmadha Rao and Maggie Paulose and Ram Neupane and Robert Vajtai and Ching-Wu Chu and Gelu Costin and Douglas S. Galvao and Angel A. Marti and Peter van Aken and Oomman K Varghese and Chandra Sekhar Tiwary and M R Anantharaman and Pulickel M Ajayan
},
url = {https://www.nature.com/articles/s41565-018-0134-y},
year = {2018},
date = {2018-05-07},
journal = {Nature Nanotechnology},
volume = {13},
pages = {602--610},
abstract = {With the advent of graphene, the most studied of all two-dimensional materials, many inorganic analogues have been synthesized and are being exploited for novel applications. Several approaches have been used to obtain large-grain, high-quality materials. Naturally occurring ores, for example, are the best precursors for obtaining highly ordered and large-grain atomic layers by exfoliation. Here, we demonstrate a new two-dimensional material ‘hematene’ obtained from natural iron ore hematite (α-Fe2O3), which is isolated by means of liquid exfoliation. The two-dimensional morphology of hematene is confirmed by transmission electron microscopy. Magnetic measurements together with density functional theory calculations confirm the ferromagnetic order in hematene while its parent form exhibits antiferromagnetic order. When loaded on titania nanotube arrays, hematene exhibits enhanced visible light photocatalytic activity. Our study indicates that photogenerated electrons can be transferred from hematene to titania despite a band alignment unfavourable for charge transfer.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Eliezer F.; Autreto Oliveira, Pedro A. S. ; Woellner
On the mechanical properties of novamene: a fully atomistic molecular dynamics and DFT investigation Online
2018, (preprint ArXiv:1804.07215).
@online{Oliveira2018f,
title = {On the mechanical properties of novamene: a fully atomistic molecular dynamics and DFT investigation},
author = {Oliveira, Eliezer F.; Autreto, Pedro A. S.; Woellner, Cristiano F.; Galvao, Douglas S.},
url = {https://arxiv.org/abs/1804.07215},
year = {2018},
date = {2018-04-19},
abstract = {We have investigated through fully atomistic reactive molecular dynamics and DFT simulations, the mechanical properties and fracture dynamics of novamene, a new 3D carbon allotrope structure recently proposed. Our results showed that novamene is an anisotropic structure with relation to tensile deformation. Although novamente shares some mechanical features with other carbon allotropes, it also exhibits distinct ones, such as, extensive structural reconstructions (self-healing effect). Novamene presents ultimate strength (~ 100 GPa) values lower than other carbon allotropes, but it has the highest ultimate strain along the z-direction (~ 22.5%). Although the Young's modulus (~ 600 GPa) and ultimate strength values are smaller than for other carbon allotropes, they still outperform other materials, such as for example silicon, steel or titanium alloys. With relation to the fracture dynamics, novamene is again anisotropic with the fracture/crack propagation originating from deformed heptagons and pentagons for x and y directions and broken sp3 bonds connecting structural planes. Another interesting feature is the formation of multiple and long carbon linear chains in the final fracture stages.},
note = {preprint ArXiv:1804.07215},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Kabbani, Mohamad A.; Kochat, Vidya; Bhowmick, Sanjit; Soto, Matias; Som, Anirban; Krishnadas, K. R.; Woellner, Cristiano F.; Jaques, Ygor M.; Barrera, Enrique V.; Asif, Syed; Vajtai, Robert; Pradeep, Thalappil; Galvão, Douglas S.; Kabbani, Ahmad T.; Tiwary, Chandra Sekhar; Ajayan, Pulickel M.
Consolidation of Functionalized Graphene at Ambient Temperature via Mechano-chemistry Journal Article
Em: Carbon, vol. 134, não 8, pp. 491-499, 2018.
@article{Kabbani2018,
title = {Consolidation of Functionalized Graphene at Ambient Temperature via Mechano-chemistry},
author = {Mohamad A. Kabbani and Vidya Kochat and Sanjit Bhowmick and Matias Soto and Anirban Som and K.R. Krishnadas and Cristiano F. Woellner and Ygor M. Jaques and Enrique V. Barrera and Syed Asif and Robert Vajtai and Thalappil Pradeep and Douglas S. Galvão and Ahmad T. Kabbani and Chandra Sekhar Tiwary and Pulickel M. Ajayan},
url = {https://www.sciencedirect.com/science/article/pii/S0008622318302987?dgcid=raven_sd_aip_email},
doi = {DOI:10.1016/j.carbon.2018.03.049},
year = {2018},
date = {2018-03-22},
journal = {Carbon},
volume = {134},
number = {8},
pages = {491-499},
abstract = {Graphitic solids are typically produced via high temperature and energy consuming
processing (e.g. sintering) of carbon particles. Here, we demonstrate the mechano-chemical
assembly of functionalized graphene layers into 3D graphitic solids via room temperature and
low energy consuming processing. The chemical functional groups on graphene layers are
interconnected at room temperature under pressure leading to porous three-dimensional
structures with tunable mechanical and electrical properties. The formation of mechanochemistry
induced atomic scale junctions and their impact on mechanical properties of
graphene assembled carbon materials are demonstrated through nano-indentation experiments
and confirmed using DFT and molecular dynamics simulations. The results show room
temperature consolidation routes of graphene layers into bulk carbon solids.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
processing (e.g. sintering) of carbon particles. Here, we demonstrate the mechano-chemical
assembly of functionalized graphene layers into 3D graphitic solids via room temperature and
low energy consuming processing. The chemical functional groups on graphene layers are
interconnected at room temperature under pressure leading to porous three-dimensional
structures with tunable mechanical and electrical properties. The formation of mechanochemistry
induced atomic scale junctions and their impact on mechanical properties of
graphene assembled carbon materials are demonstrated through nano-indentation experiments
and confirmed using DFT and molecular dynamics simulations. The results show room
temperature consolidation routes of graphene layers into bulk carbon solids.
Han, Yang; Zhou, Yanguang; Qin, Guangzhao; Dong, Jinming; Galvao, Douglas S; Hu, Ming
Unprecedented mechanical response of the lattice thermal conductivity of auxetic carbon crystals Journal Article
Em: Carbon, vol. 122, pp. 374-380, 2017.
@article{Han2017,
title = {Unprecedented mechanical response of the lattice thermal conductivity of auxetic carbon crystals},
author = {Han, Yang and Zhou, Yanguang and Qin, Guangzhao and Dong, Jinming and Galvao, Douglas S and Hu, Ming},
url = {http://www.sciencedirect.com/science/article/pii/S0008622317306760},
doi = {10.1016/j.carbon.2017.06.100},
year = {2017},
date = {2017-10-01},
journal = {Carbon},
volume = {122},
pages = {374-380},
abstract = {Lattice thermal conductivity (κ) of bulk materials usually increases under compression and decreases under tension, while there are still some unusual systems, exhibiting reduced κ when compressed. However, to date it has never been reported for a bulk material, whose κ is substantially enhanced under tensile strain. In this paper, we have studied thermal transport of three auxetic carbon crystals: cis-C, trans-C and hin-C for short, and their strain responses by performing first-principles calculations. It is intriguing to find that their κ are much lower than those of their allotropes, and further decrease abnormally under compression. More strikingly, κ of trans-C (cis-C) anomalously increases with tensile strain up to 7% (6%) with maximum κ of almost 7 (5) times larger than the unstrained value. The abnormal strain dependent κ are attributed to the dominant role of the enhancement of phonon lifetime under stretching, which can be further explained from the unique atomic structure of the main chain of polydiacetylene in trans-C and cis-C. The weakening of phonon anharmonicity is reflected by the enhancement of root mean-square displacement values. The reported giant augmentation of κ may inspire intensive research on auxetic carbon crystals as potential materials for emerging nanoelectronic devices.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
de Sousa, JM; Aguiar, AL; Girao, EC; Fonseca, Alexandre F; AG Filho, Souza; Galvao, Douglas S
Mechanical Properties and Fracture Patterns of Pentagraphene Membranes Online
2017, (preprint arXiv:1703.03789).
@online{deSousa2017,
title = {Mechanical Properties and Fracture Patterns of Pentagraphene Membranes},
author = {de Sousa, JM and Aguiar, AL and Girao, EC and Fonseca, Alexandre F and AG Filho, Souza and Galvao, Douglas S},
url = {https://arxiv.org/abs/1703.03789},
year = {2017},
date = {2017-03-10},
abstract = {Recently, a new two-dimensional carbon allotrope called pentagraphene (PG) was
proposed. PG exhibits mechanical and electronic interesting properties, including typical
band gap values of semiconducting materials. PG has a Cairo-tiling-like 2D lattice
of non coplanar pentagons and its mechanical properties have not been yet fully investigated.
In this work, we combined density functional theory (DFT) calculations and
reactive molecular dynamics (MD) simulations to investigate the mechanical properties
and fracture patterns of PG membranes under tensile strain. We show that PG
membranes can hold up to 20% of strain and that fracture occurs only after substantial
dynamical bond breaking and the formation of 7, 8 and 11 carbon rings and carbon
chains. The stress-strain behavior was observed to follow two regimes, one exhibiting linear elasticity followed by a plastic one, involving carbon atom re-hybridization with
the formation of carbon rings and chains. Our results also show that mechanically
induced structural transitions from PG to graphene is unlikely to occur, in contrast to
what was previously speculated in the literature.},
note = {preprint arXiv:1703.03789},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
proposed. PG exhibits mechanical and electronic interesting properties, including typical
band gap values of semiconducting materials. PG has a Cairo-tiling-like 2D lattice
of non coplanar pentagons and its mechanical properties have not been yet fully investigated.
In this work, we combined density functional theory (DFT) calculations and
reactive molecular dynamics (MD) simulations to investigate the mechanical properties
and fracture patterns of PG membranes under tensile strain. We show that PG
membranes can hold up to 20% of strain and that fracture occurs only after substantial
dynamical bond breaking and the formation of 7, 8 and 11 carbon rings and carbon
chains. The stress-strain behavior was observed to follow two regimes, one exhibiting linear elasticity followed by a plastic one, involving carbon atom re-hybridization with
the formation of carbon rings and chains. Our results also show that mechanically
induced structural transitions from PG to graphene is unlikely to occur, in contrast to
what was previously speculated in the literature.
Borges, Daiane Damasceno; Maurin, Guillaume; Galvao, Douglas S
Design of Porous Metal-Organic Frameworks for Adsorption Driven Thermal Batteries Journal Article
Em: MRS Advances, vol. 2017, pp. 1-6, 2017.
@article{Borges2017b,
title = {Design of Porous Metal-Organic Frameworks for Adsorption Driven Thermal Batteries},
author = {Borges, Daiane Damasceno and Maurin, Guillaume and Galvao, Douglas S},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/design-of-porous-metalorganic-frameworks-for-adsorption-driven-thermal-batteries/A63B92E4D7E413D7CC047E152C7F22AF},
doi = {10.1557/adv.2017.181},
year = {2017},
date = {2017-02-15},
journal = {MRS Advances},
volume = {2017},
pages = {1-6},
abstract = {Thermal batteries based on a reversible adsorption/desorption of a working fluid (water, methanol, ammonia) rather than the conventional vapor compression is a promising alternative to exploit waste thermal energy for heat reallocation. In this context, there is an increasing interest to find novel porous solids able to adsorb a high energy density of working fluid under low relative vapor pressure condition combined with an easy ability of regeneration (desorption) at low temperature, which are the major requirements for adsorption driven heat pumps and chillers. The porous crystalline hybrid materials named Metal–Organic Frameworks (MOF) represent a great source of inspiration for sorption based-applications owing to their tunable chemical and topological features associated with a large variability of pore sizes. Recently, we have designed a new MOF named MIL-160 (MIL stands for Materials of Institut Lavoisier), isostructural to CAU-10, built from the assembly of corner sharing aluminum chains octahedra AlO4(OH)2 with the 2,5-furandicarboxylic linker substituting the pristine organic linker, 1,4-benzenedicarboxylate. This ligand replacement strategy proved to enhance both the hydrophilicity of the MOF and its amount of water adsorbed at low p/p0. This designed solid was synthesized and its chemical stability/adsorption performances verified. Here, we have extended this study by incorporating other polar heterocyclic linkers and a comparative computational study of the water adsorption performances of these novel structures has been performed. To that purpose, the cell and geometry optimizations of all hypothetical frameworks were first performed at the density functional theory level and their water adsorption isotherms were further predicted by using force-field based Grand-Canonical Monte Carlo simulations. This study reveals the ease tunable water affinity of MOF for the desired application.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Chandra Sekhar Tiwary Sujin P Jose, Suppanat Kosolwattana
Enhanced supercapacitor performance of a 3D architecture tailored using atomically thin rGO–MoS 2 2D sheets Journal Article
Em: RSC Advances, vol. 6, pp. 93384-93393, 2016.
@article{Jose2016,
title = {Enhanced supercapacitor performance of a 3D architecture tailored using atomically thin rGO–MoS 2 2D sheets},
author = {Sujin P Jose, Chandra Sekhar Tiwary, Suppanat Kosolwattana, Prasanth Raghavan, Leonardo D Machado, Chandkiram Gautam, T Prasankumar, Jarin Joyner, Sehmus Ozden, Douglas S Galvao, PM Ajayan},
url = {xlink.rsc.org/?DOI=c6ra20960b},
doi = {10.1039/C6RA20960B},
year = {2016},
date = {2016-09-19},
journal = {RSC Advances},
volume = {6},
pages = {93384-93393},
abstract = {A 3D architecture is fabricated using 2D nano-sheets of GO and MoS2 as the building blocks by a facile, one-pot chronoamperometry method to achieve a conductive additive free, binder free and scalable supercapacitor electrode. The superior electrochemical properties of the 3D PPy-rGO–MoS2 (PGMo) are due to its porous structure, thin wall, high surface area and high electrical conductivity that endow rapid transportation of electrolyte ions and electrons throughout the electrode matrix. The synergistic effect between the components in a proper ratio improves the supercapacitor performance and material stability of PGMo. The possible correlation of the structure and electrochemical performance of the 3D ternary composite is backed by a fully atomistic molecular dynamics (MD) simulation study. The high specific capacitance (387 F g−1) and impressive cycling stability (>1000 cycles) estimated for PGMo open up an opportunity to consider the 3D ternary nanostructures as cutting edge materials for energy storage solutions.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Chandra Sekhar Tiwary Mohamad A Kabbani, Anirban Som
A generic approach for mechano-chemical reactions between carbon nanotubes of different functionalities Journal Article
Em: Carbon, vol. 104, pp. 196-202, 2016.
@article{Kabbani2016,
title = {A generic approach for mechano-chemical reactions between carbon nanotubes of different functionalities},
author = {Mohamad A Kabbani, Chandra Sekhar Tiwary, Anirban Som, KR Krishnadas, Pedro AS Autreto, Sehmus Ozden, Kunttal Keyshar, Ken Hackenberg, Alin Christian Chipara, Douglas S Galvao, Robert Vajtai, Ahmad T Kabbani, Thalappil Pradeep, Pulickel M Ajayan},
url = {www.sciencedirect.com/science/article/pii/S000862231630183X},
doi = {10.1016/j.carbon.2016.02.094},
year = {2016},
date = {2016-08-31},
journal = {Carbon},
volume = {104},
pages = {196-202},
abstract = {Abstract Here, we report similar reactions between nanotubes carrying functionalities,
namely carbon nanotubes (CNTs) with the acyl chloride/hydroxyl and amine/carboxylic
functionalities directly attached to their surfaces, resulting in the formation ofchemically
modified graphene products. The reaction is spontaneous and is facilitated by simple
grinding of the reactants. The new solid-state reactions have been confirmed using different
spectroscopic and electron microscopy techniques.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
namely carbon nanotubes (CNTs) with the acyl chloride/hydroxyl and amine/carboxylic
functionalities directly attached to their surfaces, resulting in the formation ofchemically
modified graphene products. The reaction is spontaneous and is facilitated by simple
grinding of the reactants. The new solid-state reactions have been confirmed using different
spectroscopic and electron microscopy techniques.
Yongji Gong Bo Li, Zhili Hu
Solid–Vapor Reaction Growth of Transition‐Metal Dichalcogenide Monolayers Journal Article
Em: Angewandte Chemie, vol. 128, não 36, pp. 10814-10819, 2016.
@article{Li2016,
title = {Solid–Vapor Reaction Growth of Transition‐Metal Dichalcogenide Monolayers},
author = {Bo Li, Yongji Gong, Zhili Hu, Gustavo Brunetto, Yingchao Yang, Gonglan Ye, Zhuhua Zhang, Sidong Lei, Zehua Jin, Elisabeth Bianco, Xiang Zhang, Weipeng Wang, Jun Lou, Douglas S Galvão, Ming Tang, Boris I Yakobson, Robert Vajtai, Pulickel M Ajayan},
url = {onlinelibrary.wiley.com/doi/10.1002/anie.201604445/abstract},
doi = {10.1002/ange.201604445},
year = {2016},
date = {2016-08-26},
journal = {Angewandte Chemie},
volume = {128},
number = {36},
pages = {10814-10819},
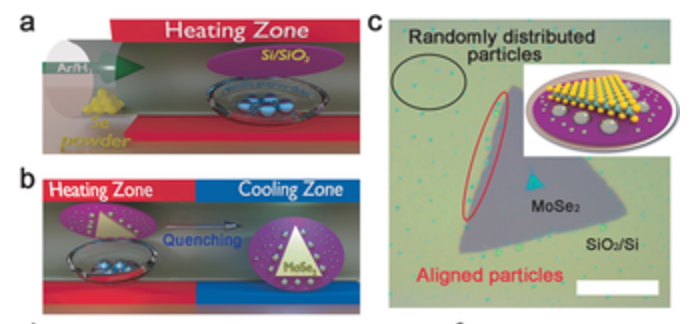
abstract = {Two-dimensional (2D) layered semiconducting transition-metal dichalcogenides (TMDCs) are promising candidates for next-generation ultrathin, flexible, and transparent electronics. Chemical vapor deposition (CVD) is a promising method for their controllable, scalable synthesis but the growth mechanism is poorly understood. Herein, we present systematic studies to understand the CVD growth mechanism of monolayer MoSe2, showing reaction pathways for growth from solid and vapor precursors. Examination of metastable nanoparticles deposited on the substrate during growth shows intermediate growth stages and conversion of non-stoichiometric nanoparticles into stoichiometric 2D MoSe2 monolayers. The growth steps involve the evaporation and reduction of MoO3 solid precursors to sub-oxides and stepwise reactions with Se vapor to finally form MoSe2. The experimental results and proposed model were corroborated by ab initio Car–Parrinello molecular dynamics studies.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Rodrigo Prioli Clara M Almeida, Benjamin Fragneaud
Giant and Tunable Anisotropy of Nanoscale Friction in Graphene Journal Article
Em: Nature Scientific Reports, vol. 6, pp. 31569, 2016.
@article{Almeida2016,
title = {Giant and Tunable Anisotropy of Nanoscale Friction in Graphene},
author = {Clara M Almeida, Rodrigo Prioli, Benjamin Fragneaud, Luiz Gustavo Cançado, Ricardo Paupitz, Douglas S Galvão, Marcelo De Cicco, Marcos G Menezes, Carlos A Achete, Rodrigo B Capaz},
url = {http://www-nature-com.ez88.periodicos.capes.gov.br/articles/srep31569},
doi = {10.1038/srep31569},
year = {2016},
date = {2016-07-18},
journal = {Nature Scientific Reports},
volume = {6},
pages = {31569},
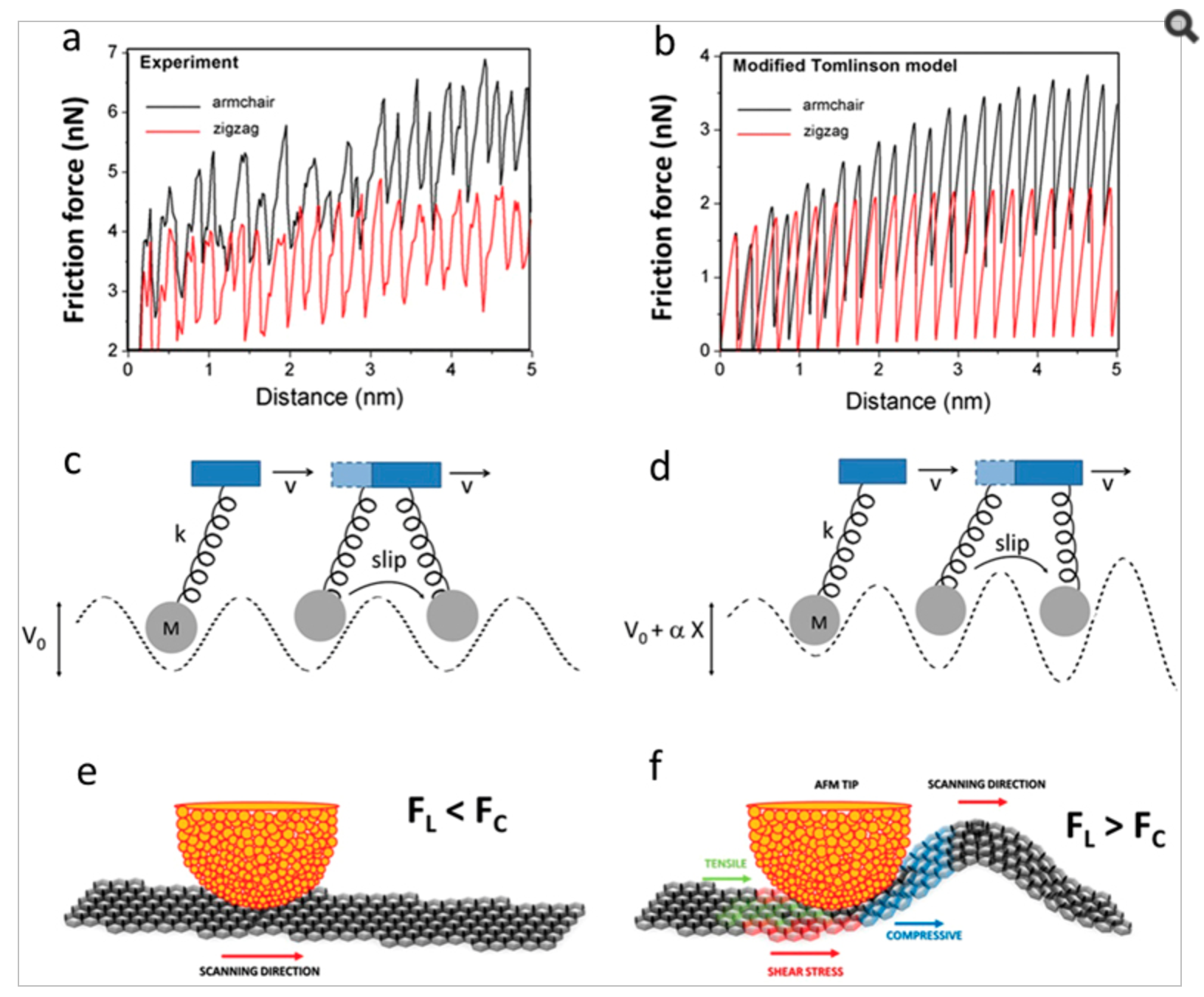
abstract = {The nanoscale friction between an atomic force microscopy tip and graphene is investigated using friction force microscopy (FFM). During the tip movement, friction forces are observed to increase and then saturate in a highly anisotropic manner. As a result, the friction forces in graphene are highly dependent on the scanning direction: under some conditions, the energy dissipated along the armchair direction can be 80% higher than along the zigzag direction. In comparison, for highly-oriented pyrolitic graphite (HOPG), the friction anisotropy between armchair and zigzag directions is only 15%. This giant friction anisotropy in graphene results from anisotropies in the amplitudes of flexural deformations of the graphene sheet driven by the tip movement, not present in HOPG. The effect can be seen as a novel manifestation of the classical phenomenon of Euler buckling at the nanoscale, which provides the non-linear ingredients that amplify friction anisotropy. Simulations based on a novel version of the 2D Tomlinson model (modified to include the effects of flexural deformations), as well as fully atomistic molecular dynamics simulations and first-principles density-functional theory (DFT) calculations, are able to reproduce and explain the experimental observations.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Gao, Guanhui; Mathkar, Akshay; Martins, Eric Perim; Galvao, Douglas S; Gao, Duyang; da Silva Autreto, Pedro Alves; Sun, Chengjun; Cai, Lintao; Ajayan, Pulickel M
Designing nanoscaled hybrids from atomic layered boron nitride with silver nanoparticle deposition Journal Article
Em: Journal of Materials Chemistry A, vol. 2, não 9, pp. 3148–3154, 2014.
@article{gao2014designing,
title = {Designing nanoscaled hybrids from atomic layered boron nitride with silver nanoparticle deposition},
author = {Gao, Guanhui and Mathkar, Akshay and Martins, Eric Perim and Galvao, Douglas S and Gao, Duyang and da Silva Autreto, Pedro Alves and Sun, Chengjun and Cai, Lintao and Ajayan, Pulickel M},
url = {http://pubs.rsc.org/en/Content/ArticleLanding/2014/TA/c3ta12892j#!divAbstract},
year = {2014},
date = {2014-01-01},
journal = {Journal of Materials Chemistry A},
volume = {2},
number = {9},
pages = {3148--3154},
publisher = {Royal Society of Chemistry},
abstract = {We have developed a microwave assisted one-pot approach to fabricate a novel hybrid nano-composite composed of two-dimensional chemically exfoliated layered hexagonal boron nitride (h-BN) and embedded silver nanoparticles (SNP). Atomic layered h-BN exfoliated using chemical liquid showed strong in-plane bonding and weak van der Waals interplanar interactions, which is utilized for chemically interfacing SNP, indicating their ability to act as excellent nano-scaffolds. The SNP/h-BN optical response, in particular band gap, is strongly dependent on the concentration of the metallic particles. In order to gain further insight into this behavior we have also carried out ab initio density functional theory (DFT) calculations on modeled structures, demonstrating that the bandgap value of SNP/h-BN hybrids could be significantly altered by a small percentage of OH− groups located at dangling B and N atoms. Our results showed that these novel SNP/h-BN nanohybrid structures exhibited excellent thermal stability and they are expected to be applied as devices for thermal oxidation-resistant surface enhanced Raman spectroscopy (SERS). The SNP/h-BN membrane showed remarkable antibacterial activity, suggesting their potential use in water disinfection and food packaging.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Perim, E; Paupitz, R; Botari, T; Galvao, DS
One-dimensional silicon and germanium nanostructures with no carbon analogues Journal Article
Em: Physical Chemistry Chemical Physics, vol. 16, não 44, pp. 24570–24574, 2014.
@article{perim2014one,
title = {One-dimensional silicon and germanium nanostructures with no carbon analogues},
author = {Perim, E and Paupitz, R and Botari, T and Galvao, DS},
url = {http://pubs.rsc.org/en/content/articlehtml/2014/cp/c4cp03708a},
year = {2014},
date = {2014-01-01},
journal = {Physical Chemistry Chemical Physics},
volume = {16},
number = {44},
pages = {24570--24574},
publisher = {Royal Society of Chemistry},
abstract = {In this work we report new silicon and germanium tubular nanostructures with no corresponding stable carbon analogues. The electronic and mechanical properties of these new tubes were investigated through ab initio methods. Our results show that these structures have lower energy than their corresponding nanoribbon structures and are stable up to high temperatures (500 and 1000 K, for silicon and germanium tubes, respectively). Both tubes are semiconducting with small indirect band gaps, which can be significantly altered by both compressive and tensile strains. Large bandgap variations of almost 50% were observed for strain rates as small as 3%, suggesting their possible applications in sensor devices. They also present high Young's modulus values (0.25 and 0.15 TPa, respectively). TEM images were simulated to help in the identification of these new structures.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Perim, Eric; Paupitz, Ricardo; Botari, Tiago; Galvao, Douglas S
Novel Semiconducting Silicon and Germanium Nanotubes Journal Article
Em: arXiv preprint arXiv:1403.2061, 2014.
@article{perim2014novel,
title = {Novel Semiconducting Silicon and Germanium Nanotubes},
author = {Perim, Eric and Paupitz, Ricardo and Botari, Tiago and Galvao, Douglas S},
url = {http://arxiv.org/abs/1403.2061},
year = {2014},
date = {2014-01-01},
journal = {arXiv preprint arXiv:1403.2061},
abstract = {In this work we report new silicon and germanium nanotube structures, with no corresponding
stable carbon analogues and which cannot be described by integer chiral indices. The electronic
and mechanical properties of these new tubes were investigated through ab initio methods. Our
results show that the structures are stable up to high temperatures (500 and 1000 K, for silicon and
germanium tubes, respectively). Both tubes are semiconducting with small indirect band gaps,
which can be significantly altered by both compressive and tensile strains. They also present high
Young modulus values (0.25 and 0.15 TPa, respectively).},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
stable carbon analogues and which cannot be described by integer chiral indices. The electronic
and mechanical properties of these new tubes were investigated through ab initio methods. Our
results show that the structures are stable up to high temperatures (500 and 1000 K, for silicon and
germanium tubes, respectively). Both tubes are semiconducting with small indirect band gaps,
which can be significantly altered by both compressive and tensile strains. They also present high
Young modulus values (0.25 and 0.15 TPa, respectively).
Autreto, Pedro Alves da Silva; Galvao, Douglas S; Artacho, Emilio
Species Fractionation in Atomic Chains from Mechanically Stretched Alloys Journal Article
Em: arXiv preprint arXiv:1312.1285, 2013.
@article{autreto2013species,
title = {Species Fractionation in Atomic Chains from Mechanically Stretched Alloys},
author = {Autreto, Pedro Alves da Silva and Galvao, Douglas S and Artacho, Emilio},
url = {http://arxiv.org/abs/1312.1285},
year = {2013},
date = {2013-01-01},
journal = {arXiv preprint arXiv:1312.1285},
abstract = {Bettini et al. [Nature Nanotech 1, 182 (2006)] reported the first experimental realization of linear
atomic chains (LACs) composed of different atoms (Au and Ag). Different contents of Au and Ag
were observed in the chains from what found in the bulk alloys, which rises the question of what is the
wire composition if in equilibrium with a bulk alloy. In this work we address the thermodynamic
driving force for species fractionation in LACs under tension, and we present density-functional
theory results for Ag-Au chain alloys. A pronounced stabilization of wires with an alternating
Ag-Au sequence is observed, which could be behind the experimentally observed Au enrichment in
LACs from alloys of high Ag content.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
atomic chains (LACs) composed of different atoms (Au and Ag). Different contents of Au and Ag
were observed in the chains from what found in the bulk alloys, which rises the question of what is the
wire composition if in equilibrium with a bulk alloy. In this work we address the thermodynamic
driving force for species fractionation in LACs under tension, and we present density-functional
theory results for Ag-Au chain alloys. A pronounced stabilization of wires with an alternating
Ag-Au sequence is observed, which could be behind the experimentally observed Au enrichment in
LACs from alloys of high Ag content.
Brunetto, Gustavo; Autreto, PAS; Machado, Leonardo Dantas; Santos, BI; dos Santos, Ricardo PB; Galvao, Douglas S
Nonzero gap two-dimensional carbon allotrope from porous graphene Journal Article
Em: The Journal of Physical Chemistry C, vol. 116, não 23, pp. 12810–12813, 2012.
@article{brunetto2012nonzero,
title = {Nonzero gap two-dimensional carbon allotrope from porous graphene},
author = {Brunetto, Gustavo and Autreto, PAS and Machado, Leonardo Dantas and Santos, BI and dos Santos, Ricardo PB and Galvao, Douglas S},
url = {http://pubs.acs.org/doi/abs/10.1021/jp211300n},
year = {2012},
date = {2012-01-01},
journal = {The Journal of Physical Chemistry C},
volume = {116},
number = {23},
pages = {12810--12813},
publisher = {American Chemical Society},
abstract = {Graphene is considered one of the most promising materials for future electronics. However, in its pristine form, graphene is a gapless material, which imposes limitations to its use in some electronic applications. To solve this problem, many approaches have been tried, such as physical and chemical functionalizations. These processes compromise some of the desirable graphene properties. In this work, based on ab initio quantum molecular dynamics, we showed that a two-dimensional carbon allotrope, named biphenylene carbon (BPC), can be obtained from selective dehydrogenation of porous graphene. BPC presents a nonzero bandgap and well-delocalized frontier orbitals. Synthetic routes to BPC are also addressed.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
PAS Autreto MJ Lagos, SB Legoas
Temperature effects on the occurrence of long interatomic distances in atomic chains formed from stretched gold nanowires Journal Article
Em: Nanotechnology, vol. 22, não 9, pp. 095705, 2011.
@article{Lagos2011,
title = {Temperature effects on the occurrence of long interatomic distances in atomic chains formed from stretched gold nanowires},
author = {MJ Lagos, PAS Autreto, SB Legoas, F Sato, V Rodrigues, DS Galvao, D Ugarte},
url = {http://iopscience.iop.org/0957-4484/22/9/095705},
year = {2011},
date = {2011-03-04},
journal = {Nanotechnology},
volume = {22},
number = {9},
pages = {095705},
abstract = {The origin of long interatomic distances in suspended gold atomic chains formed from stretched nanowires remains the object of debate despite the large amount of theoretical and experimental work. Here, we report new atomic resolution electron microscopy observations acquired at room and liquid-nitrogen temperatures and theoretical results from ab initio quantum molecular dynamics on chain formation and stability. These new data are suggestive that the long distances are due to contamination by carbon atoms originating from the decomposition of adsorbed hydrocarbon molecules.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2019
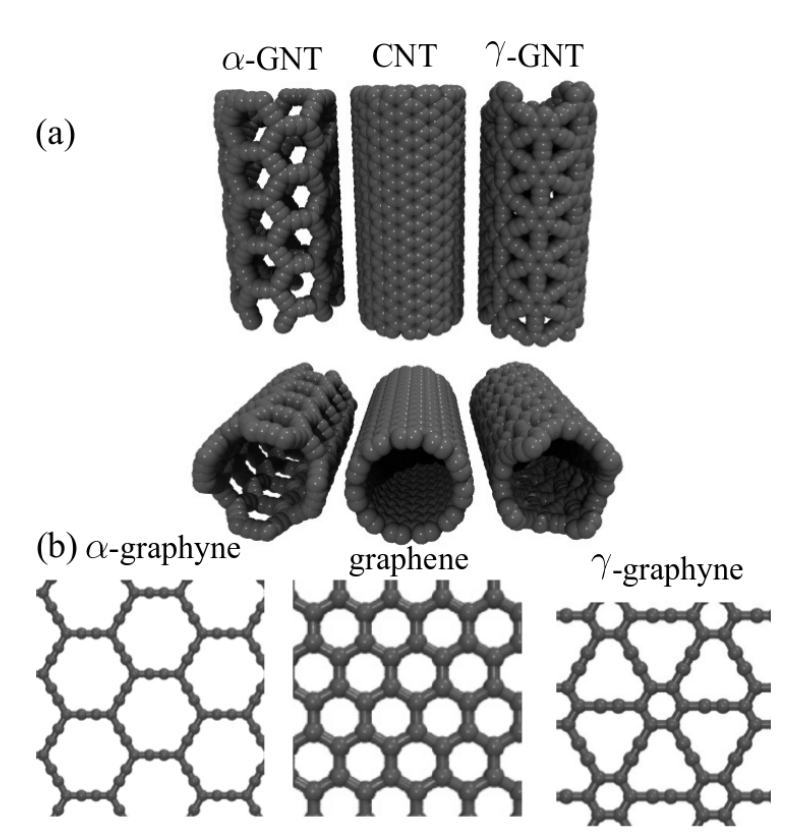
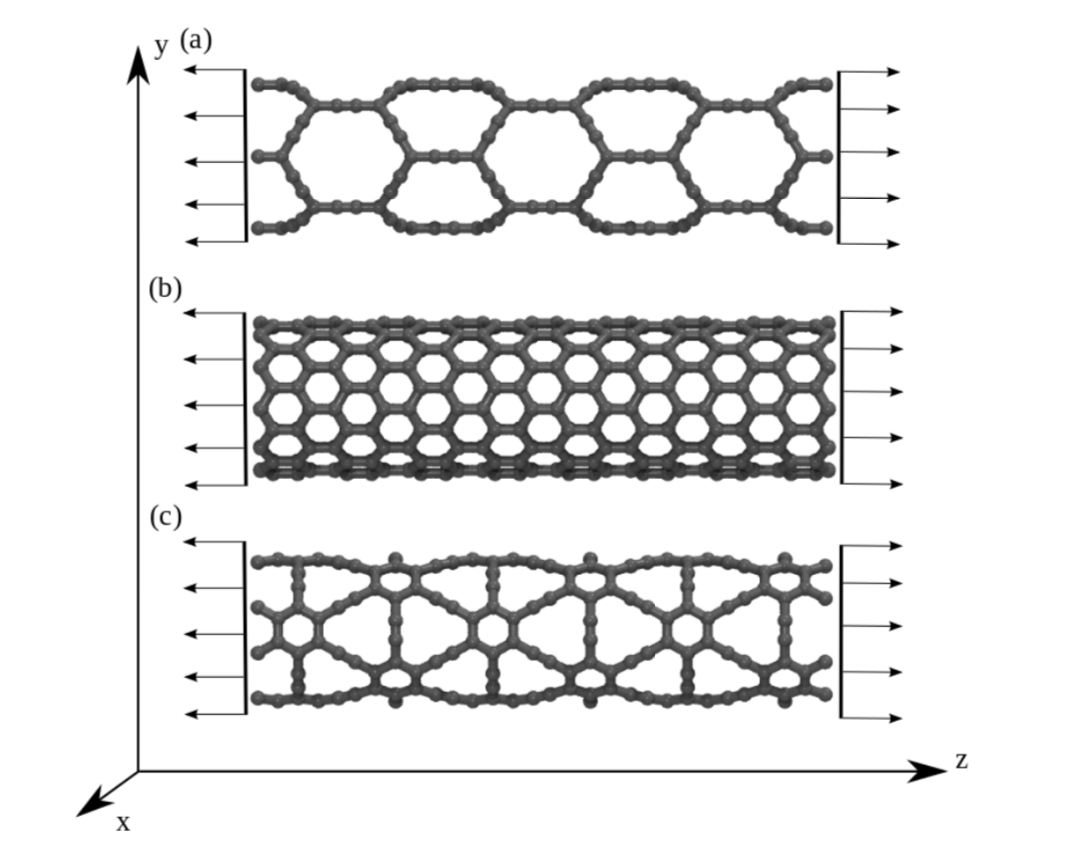
JM; Sousa, Bizao
Elastic Properties of Graphyne-based Nanotubes Online
2019, (ArXiv preprint.).
Resumo | Links | BibTeX | Tags: DFT, Graphynes, Molecular Dynamics, Nanotubes
@online{deSousa2019b,
title = {Elastic Properties of Graphyne-based Nanotubes},
author = {de Sousa, JM; , Bizao, RA; Sousa Filho, VP; Aguiar, AL; Coluci, VR; Pugno, NM; Girao, EC; Souza Filho, AG; Galvao, DS},
url = {https://arxiv.org/pdf/1905.02104.pdf},
year = {2019},
date = {2019-04-07},
abstract = {Graphyne nanotubes (GNTs) are nanostructures obtained from rolled up graphyne sheets,
in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes
are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to
conventional CNTs, GNTs can present different chiralities and electronic properties. Because
of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their
mechanical properties. In this work, we studied the mechanical response of GNTs under
tensile stress using fully atomistic molecular dynamics simulations and density functional
theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs
at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller
ultimate strength and Young’s modulus values. This is a consequence of the combined
effects of the existence of triple bonds and increased porosity/flexibility due to the presence
of acetylenic groups.},
note = {ArXiv preprint.},
keywords = {DFT, Graphynes, Molecular Dynamics, Nanotubes},
pubstate = {published},
tppubtype = {online}
}
in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes
are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to
conventional CNTs, GNTs can present different chiralities and electronic properties. Because
of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their
mechanical properties. In this work, we studied the mechanical response of GNTs under
tensile stress using fully atomistic molecular dynamics simulations and density functional
theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs
at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller
ultimate strength and Young’s modulus values. This is a consequence of the combined
effects of the existence of triple bonds and increased porosity/flexibility due to the presence
of acetylenic groups.
JM; Sousa, Bizao
Elastic Properties of Graphyne-Based Nanotubes Journal Article
Em: Computational Materials Science, vol. 170, pp. 109153, 2019.
Resumo | Links | BibTeX | Tags: DFT, Graphynes, Molecular Dynamics, Nanotubes
@article{deSousa2019c,
title = {Elastic Properties of Graphyne-Based Nanotubes},
author = {de Sousa, JM; , Bizao, RA; Sousa Filho, VP; Aguiar, AL; Coluci, VR; Pugno, NM; Girao, EC; Souza Filho, AG; Galvao, DS},
url = {https://www.sciencedirect.com/science/article/pii/S0927025619304525?dgcid=coauthor#s0040},
doi = {10.1016/j.commatsci.2019.109153},
year = {2019},
date = {2019-04-03},
journal = {Computational Materials Science},
volume = {170},
pages = {109153},
abstract = {Graphyne nanotubes (GNTs) are nanostructures obtained from rolled up graphyne sheets, in the same way carbon nanotubes (CNTs) are obtained from graphene ones. Graphynes are 2D carbon-allotropes composed of atoms in sp and sp2 hybridized states. Similarly to conventional CNTs, GNTs can present different chiralities and electronic properties. Because of the acetylenic groups (triple bonds), GNTs exhibit large sidewall pores that influence their mechanical properties. In this work, we studied the mechanical response of GNTs under tensile stress using fully atomistic molecular dynamics simulations and density functional theory (DFT) calculations. Our results show that GNTs mechanical failure (fracture) occurs at larger strain values in comparison to corresponding CNTs, but paradoxically with smaller ultimate strength and Young’s modulus values. This is a consequence of the combined effects of the existence of triple bonds and increased porosity/flexibility due to the presence of acetylenic groups.},
keywords = {DFT, Graphynes, Molecular Dynamics, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
2018
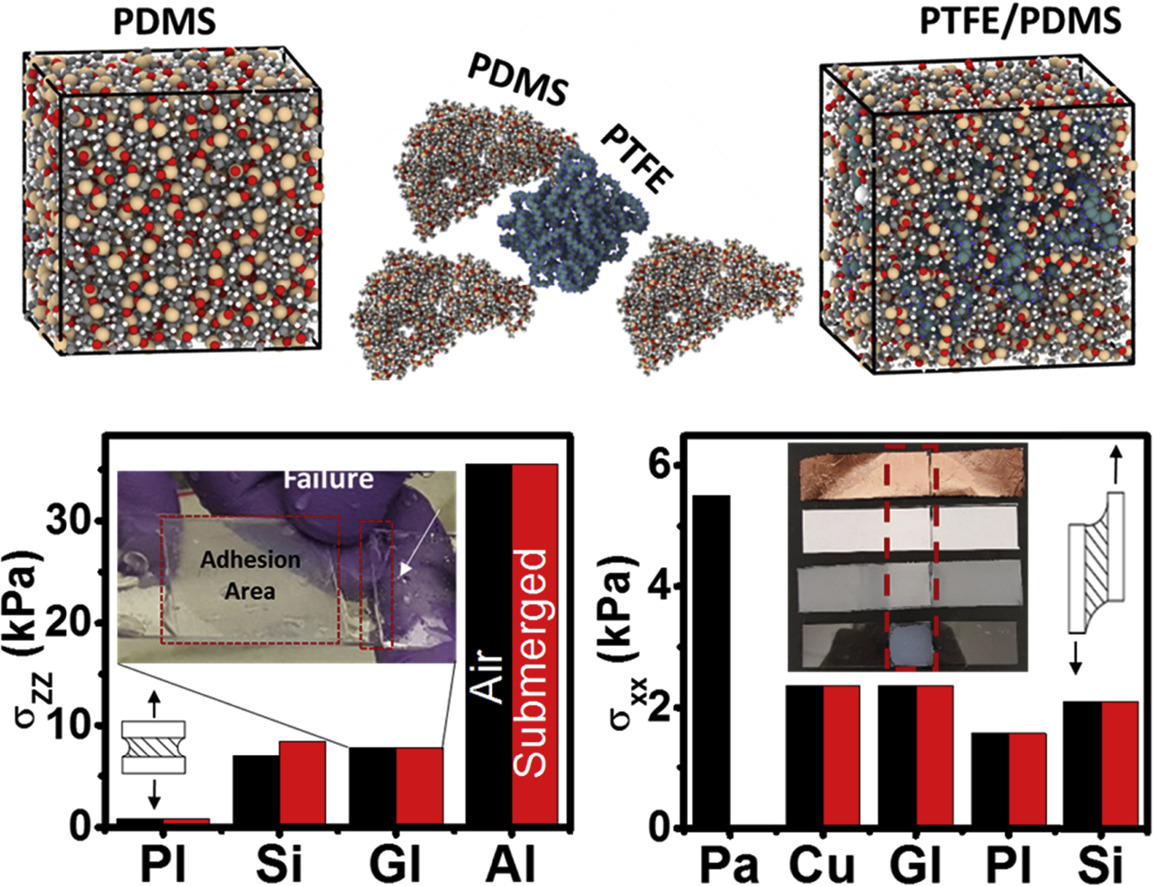
Chipara, A. C.; Tsafack, T.; Owuor, P. S.; Yeon, J.; Junkermeier, C. E.; van Duin, A. C. T.; Bhowmick, S.; Asif, S. A. S.; Radhakrishnan, S.; Park, J. H.; Brunetto, G.; Kaipparettu, B. A.; Galvão, D. S.; Chipara, M.; Lou, J.; Tsang, H. H.; Dubey, M.; Vajtai, R.; Tiwary, C. S.; Ajayan, P. M.
Underwater Adhesive using Solid–liquid Polymer Mixes Journal Article
Em: Materials Today Chemistry, vol. 9, pp. 149-157, 2018.
Resumo | Links | BibTeX | Tags: Adhesives, DFT, Molecular Dynamics, Polymer
@article{Chipara2018,
title = {Underwater Adhesive using Solid–liquid Polymer Mixes},
author = {A.C. Chipara and T. Tsafack and P.S. Owuor and J. Yeon and C.E. Junkermeier and A.C.T. van Duin and S. Bhowmick and S.A.S. Asif and S. Radhakrishnan and J.H. Park and G. Brunetto and B.A. Kaipparettu and D.S. Galvão and M. Chipara and J. Lou and H.H. Tsang and M. Dubey and R. Vajtai and C.S. Tiwary and P.M. Ajayan},
url = {https://www.sciencedirect.com/science/article/pii/S2468519418301423#appsec1},
doi = {10.1016/j.mtchem.2018.07.002},
year = {2018},
date = {2018-08-08},
journal = {Materials Today Chemistry},
volume = {9},
pages = {149-157},
abstract = {Instantaneous adhesion between different materials is a requirement for several applications ranging from electronics to biomedicine. Approaches such as surface patterning, chemical cross-linking, surface modification, and chemical synthesis have been adopted to generate temporary adhesion between various materials and surfaces. Because of the lack of curing times, temporary adhesives are instantaneous, a useful property for specific applications that need quick bonding. However, to this day, temporary adhesives have been mainly demonstrated under dry conditions and do not work well in submerged or humid environments. Furthermore, most rely on chemical bonds resulting from strong interactions with the substrate such as acrylate based. This work demonstrates the synthesis of a universal amphibious adhesive solely by combining solid polytetrafluoroethylene (PTFE) and liquid polydimethylsiloxane (PDMS) polymers. While the dipole-dipole interactions are induced by a large electronegativity difference between fluorine atoms in PTFE and hydrogen atoms in PDMS, strong surface wetting allows the proposed adhesive to fully coat both substrates and PTFE particles, thereby maximizing the interfacial chemistry. The two-phase solid–liquid polymer system displays adhesive characteristics applicable both in air and water, and enables joining of a wide range of similar and dissimilar materials (glasses, metals, ceramics, papers, and biomaterials). The adhesive exhibits excellent mechanical properties for the joints between various surfaces as observed in lap shear testing, T-peel testing, and tensile testing. The proposed biocompatible adhesive can also be reused multiple times in different dry and wet environments. Additionally, we have developed a new reactive force field parameterization and used it in our molecular dynamics simulations to validate the adhesive nature of the mixed polymer system with different surfaces. This simple amphibious adhesive could meet the need for a universal glue that performs well with a number of materials for a wide range of conditions.},
keywords = {Adhesives, DFT, Molecular Dynamics, Polymer},
pubstate = {published},
tppubtype = {article}
}
Oliveira, Eliezer F.; Autreto, Pedro A. S.; Woellner, Cristiano F.; Galvao, Douglas S.
On the mechanical properties of novamene: a fully atomistic molecular dynamics and DFT investigation Journal Article
Em: Carbon, vol. 139, pp. 782-788, 2018.
Resumo | Links | BibTeX | Tags: carbon allotropes, DFT, Molecular Dynamics, novamenes
@article{Oliveira2018e,
title = {On the mechanical properties of novamene: a fully atomistic molecular dynamics and DFT investigation},
author = {Eliezer F. Oliveira and Pedro A. S. Autreto and Cristiano F. Woellner and Douglas S. Galvao},
url = {https://www.sciencedirect.com/science/article/pii/S0008622318306882?via%3Dihub#appsec1},
doi = {10.1016/j.carbon.2018.07.038},
year = {2018},
date = {2018-07-19},
journal = {Carbon},
volume = {139},
pages = {782-788},
abstract = {We have investigated through fully atomistic reactive molecular dynamics and density functional theory simulations, the mechanical properties and fracture dynamics of single-ringed novamene (1R-novamene), a new 3D carbon allotrope structure recently proposed. Our results showed that 1R-novamene is an anisotropic structure with relation to tensile deformation. Although 1R-novamente shares some mechanical features with other carbon allotropes, it also exhibits distinct ones, such as, extensive structural reconstructions. 1R-novamene presents ultimate strength (∼100 GPa) values lower than other carbon allotropes, but it has the highest ultimate strain along the z-direction (∼22.5%). Although the Young's modulus (∼600 GPa) and ultimate strength values are smaller than for other carbon allotropes, they still outperform other materials, such as for example silicon, steel or titanium alloys. With relation to the fracture dynamics, 1R-novamene is again anisotropic with the fracture/crack propagation originating from deformed heptagons and pentagons for x and y directions and broken sp3 bonds connecting structural planes. Another interesting feature is the formation of multiple and long carbon linear chains in the final fracture stages.},
keywords = {carbon allotropes, DFT, Molecular Dynamics, novamenes},
pubstate = {published},
tppubtype = {article}
}
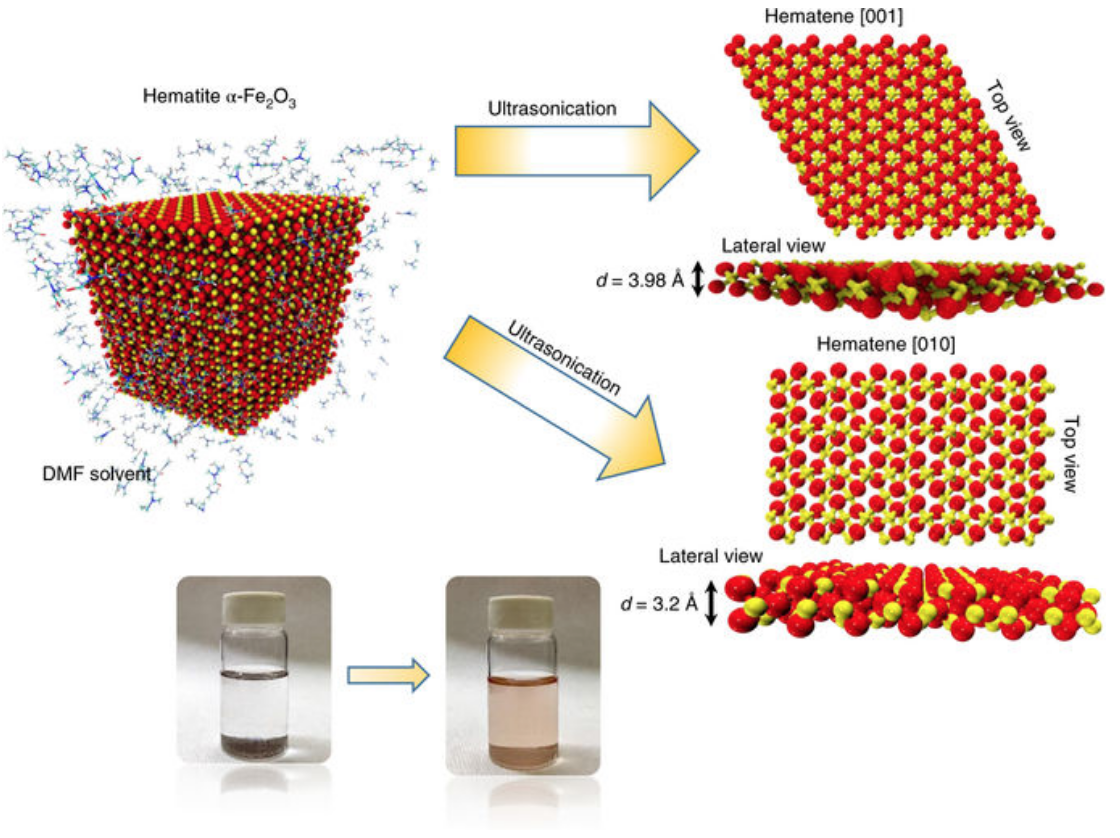
Balan, Aravind Puthirath; Radhakrishnan, Sruthi; Woellner, Cristiano F.; Sinha, Shyam K.; Deng, Liangzi; de los Reyes, Carlos; Rao, Manmadha; Paulose, Maggie; Neupane, Ram; Vajtai, Robert; Chu, Ching-Wu; Costin, Gelu; Galvao, Douglas S.; Marti, Angel A.; van Aken, Peter; Varghese, Oomman K; Tiwary, Chandra Sekhar; Anantharaman, M R; Ajayan, Pulickel M
Exfoliation of a non-van der Waals material from iron ore hematite Journal Article
Em: Nature Nanotechnology, vol. 13, pp. 602–610, 2018.
Resumo | Links | BibTeX | Tags: DFT, Hematene, Molecular Dynamics, van der Waals solids
@article{Balan2018,
title = {Exfoliation of a non-van der Waals material from iron ore hematite},
author = {Aravind Puthirath Balan and Sruthi Radhakrishnan and Cristiano F. Woellner and Shyam K. Sinha and Liangzi Deng and Carlos de los Reyes and Manmadha Rao and Maggie Paulose and Ram Neupane and Robert Vajtai and Ching-Wu Chu and Gelu Costin and Douglas S. Galvao and Angel A. Marti and Peter van Aken and Oomman K Varghese and Chandra Sekhar Tiwary and M R Anantharaman and Pulickel M Ajayan
},
url = {https://www.nature.com/articles/s41565-018-0134-y},
year = {2018},
date = {2018-05-07},
journal = {Nature Nanotechnology},
volume = {13},
pages = {602--610},
abstract = {With the advent of graphene, the most studied of all two-dimensional materials, many inorganic analogues have been synthesized and are being exploited for novel applications. Several approaches have been used to obtain large-grain, high-quality materials. Naturally occurring ores, for example, are the best precursors for obtaining highly ordered and large-grain atomic layers by exfoliation. Here, we demonstrate a new two-dimensional material ‘hematene’ obtained from natural iron ore hematite (α-Fe2O3), which is isolated by means of liquid exfoliation. The two-dimensional morphology of hematene is confirmed by transmission electron microscopy. Magnetic measurements together with density functional theory calculations confirm the ferromagnetic order in hematene while its parent form exhibits antiferromagnetic order. When loaded on titania nanotube arrays, hematene exhibits enhanced visible light photocatalytic activity. Our study indicates that photogenerated electrons can be transferred from hematene to titania despite a band alignment unfavourable for charge transfer.},
keywords = {DFT, Hematene, Molecular Dynamics, van der Waals solids},
pubstate = {published},
tppubtype = {article}
}
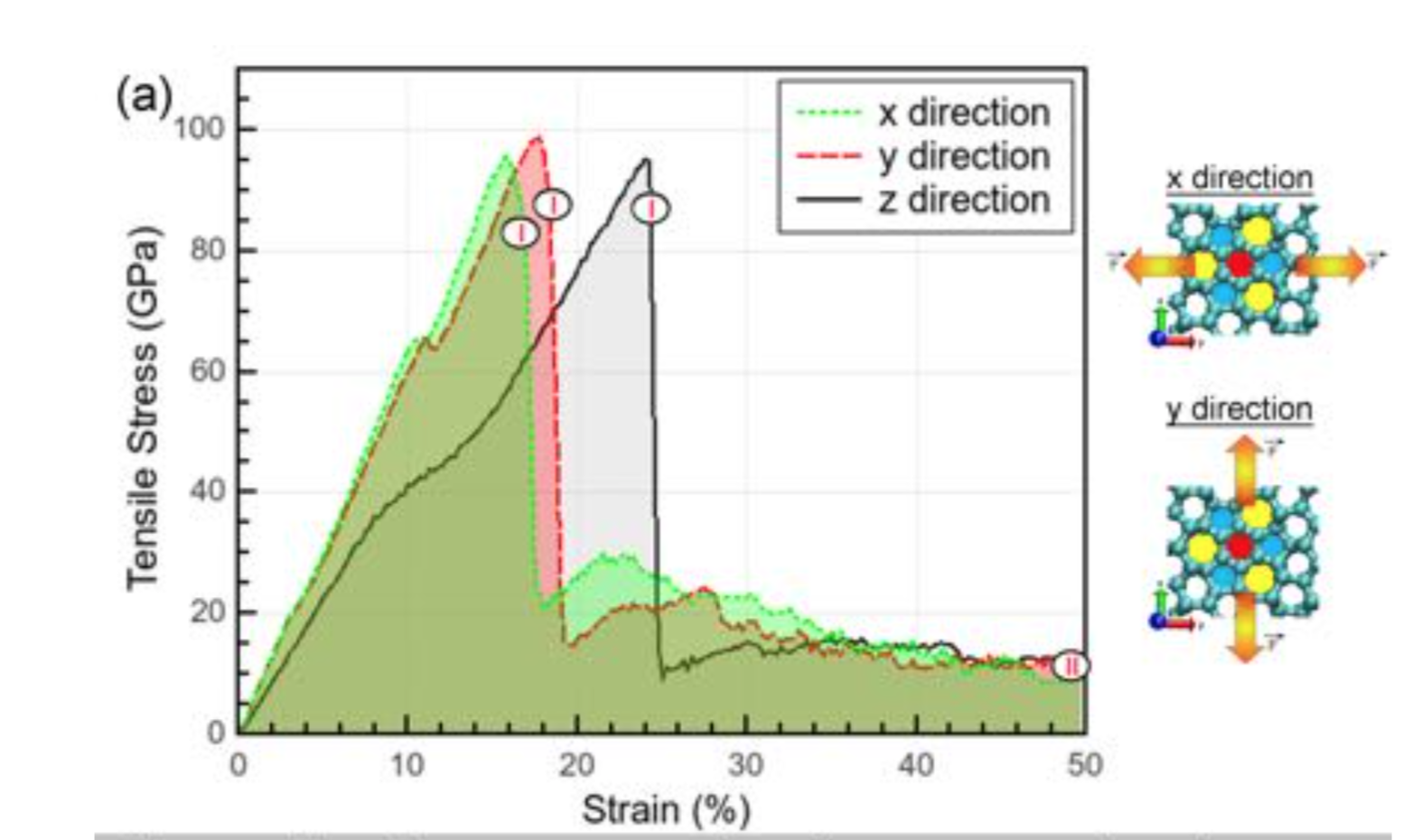
Eliezer F.; Autreto Oliveira, Pedro A. S. ; Woellner
On the mechanical properties of novamene: a fully atomistic molecular dynamics and DFT investigation Online
2018, (preprint ArXiv:1804.07215).
Resumo | Links | BibTeX | Tags: carbon allotropes, DFT, Molecular Dynamics, novamenes
@online{Oliveira2018f,
title = {On the mechanical properties of novamene: a fully atomistic molecular dynamics and DFT investigation},
author = {Oliveira, Eliezer F.; Autreto, Pedro A. S.; Woellner, Cristiano F.; Galvao, Douglas S.},
url = {https://arxiv.org/abs/1804.07215},
year = {2018},
date = {2018-04-19},
abstract = {We have investigated through fully atomistic reactive molecular dynamics and DFT simulations, the mechanical properties and fracture dynamics of novamene, a new 3D carbon allotrope structure recently proposed. Our results showed that novamene is an anisotropic structure with relation to tensile deformation. Although novamente shares some mechanical features with other carbon allotropes, it also exhibits distinct ones, such as, extensive structural reconstructions (self-healing effect). Novamene presents ultimate strength (~ 100 GPa) values lower than other carbon allotropes, but it has the highest ultimate strain along the z-direction (~ 22.5%). Although the Young's modulus (~ 600 GPa) and ultimate strength values are smaller than for other carbon allotropes, they still outperform other materials, such as for example silicon, steel or titanium alloys. With relation to the fracture dynamics, novamene is again anisotropic with the fracture/crack propagation originating from deformed heptagons and pentagons for x and y directions and broken sp3 bonds connecting structural planes. Another interesting feature is the formation of multiple and long carbon linear chains in the final fracture stages.},
note = {preprint ArXiv:1804.07215},
keywords = {carbon allotropes, DFT, Molecular Dynamics, novamenes},
pubstate = {published},
tppubtype = {online}
}
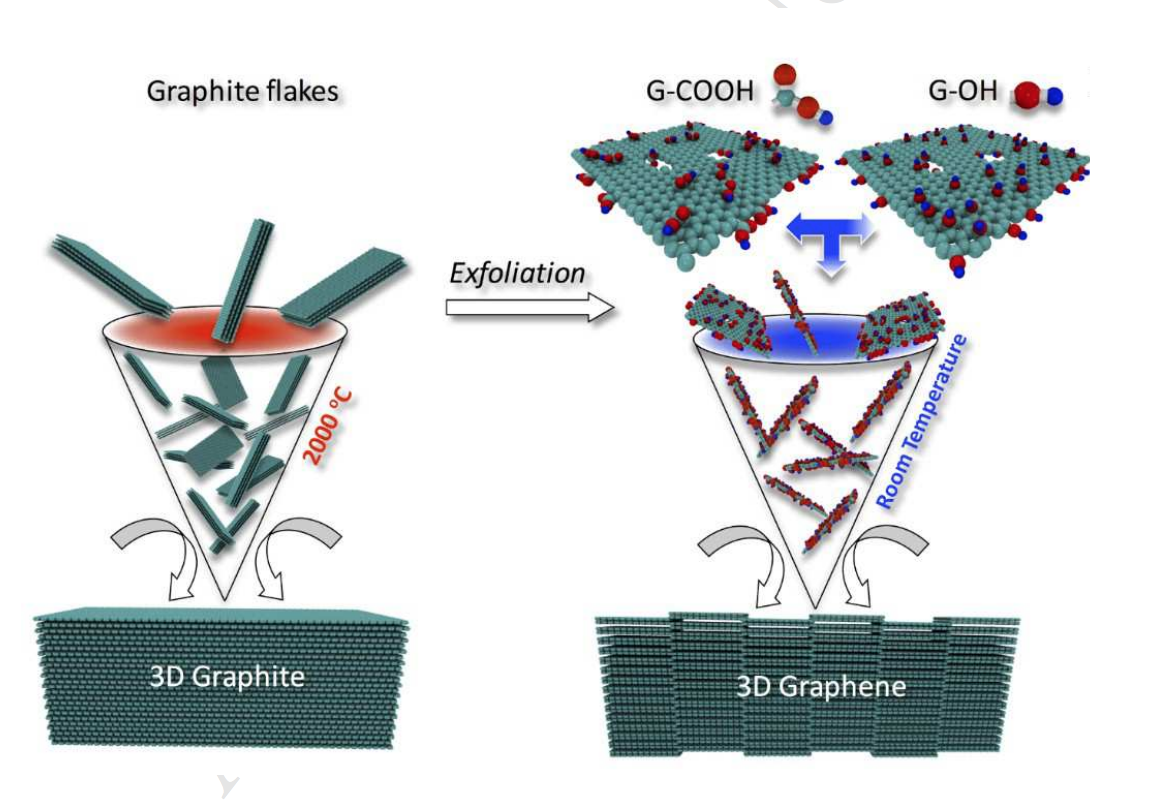
Kabbani, Mohamad A.; Kochat, Vidya; Bhowmick, Sanjit; Soto, Matias; Som, Anirban; Krishnadas, K. R.; Woellner, Cristiano F.; Jaques, Ygor M.; Barrera, Enrique V.; Asif, Syed; Vajtai, Robert; Pradeep, Thalappil; Galvão, Douglas S.; Kabbani, Ahmad T.; Tiwary, Chandra Sekhar; Ajayan, Pulickel M.
Consolidation of Functionalized Graphene at Ambient Temperature via Mechano-chemistry Journal Article
Em: Carbon, vol. 134, não 8, pp. 491-499, 2018.
Resumo | Links | BibTeX | Tags: DFT, Graphene, Mechanochemistry, Molecular Dynamics
@article{Kabbani2018,
title = {Consolidation of Functionalized Graphene at Ambient Temperature via Mechano-chemistry},
author = {Mohamad A. Kabbani and Vidya Kochat and Sanjit Bhowmick and Matias Soto and Anirban Som and K.R. Krishnadas and Cristiano F. Woellner and Ygor M. Jaques and Enrique V. Barrera and Syed Asif and Robert Vajtai and Thalappil Pradeep and Douglas S. Galvão and Ahmad T. Kabbani and Chandra Sekhar Tiwary and Pulickel M. Ajayan},
url = {https://www.sciencedirect.com/science/article/pii/S0008622318302987?dgcid=raven_sd_aip_email},
doi = {DOI:10.1016/j.carbon.2018.03.049},
year = {2018},
date = {2018-03-22},
journal = {Carbon},
volume = {134},
number = {8},
pages = {491-499},
abstract = {Graphitic solids are typically produced via high temperature and energy consuming
processing (e.g. sintering) of carbon particles. Here, we demonstrate the mechano-chemical
assembly of functionalized graphene layers into 3D graphitic solids via room temperature and
low energy consuming processing. The chemical functional groups on graphene layers are
interconnected at room temperature under pressure leading to porous three-dimensional
structures with tunable mechanical and electrical properties. The formation of mechanochemistry
induced atomic scale junctions and their impact on mechanical properties of
graphene assembled carbon materials are demonstrated through nano-indentation experiments
and confirmed using DFT and molecular dynamics simulations. The results show room
temperature consolidation routes of graphene layers into bulk carbon solids.},
keywords = {DFT, Graphene, Mechanochemistry, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
processing (e.g. sintering) of carbon particles. Here, we demonstrate the mechano-chemical
assembly of functionalized graphene layers into 3D graphitic solids via room temperature and
low energy consuming processing. The chemical functional groups on graphene layers are
interconnected at room temperature under pressure leading to porous three-dimensional
structures with tunable mechanical and electrical properties. The formation of mechanochemistry
induced atomic scale junctions and their impact on mechanical properties of
graphene assembled carbon materials are demonstrated through nano-indentation experiments
and confirmed using DFT and molecular dynamics simulations. The results show room
temperature consolidation routes of graphene layers into bulk carbon solids.
2017
Han, Yang; Zhou, Yanguang; Qin, Guangzhao; Dong, Jinming; Galvao, Douglas S; Hu, Ming
Unprecedented mechanical response of the lattice thermal conductivity of auxetic carbon crystals Journal Article
Em: Carbon, vol. 122, pp. 374-380, 2017.
Resumo | Links | BibTeX | Tags: Auxetics, DFT, Thermal, Tubulanes
@article{Han2017,
title = {Unprecedented mechanical response of the lattice thermal conductivity of auxetic carbon crystals},
author = {Han, Yang and Zhou, Yanguang and Qin, Guangzhao and Dong, Jinming and Galvao, Douglas S and Hu, Ming},
url = {http://www.sciencedirect.com/science/article/pii/S0008622317306760},
doi = {10.1016/j.carbon.2017.06.100},
year = {2017},
date = {2017-10-01},
journal = {Carbon},
volume = {122},
pages = {374-380},
abstract = {Lattice thermal conductivity (κ) of bulk materials usually increases under compression and decreases under tension, while there are still some unusual systems, exhibiting reduced κ when compressed. However, to date it has never been reported for a bulk material, whose κ is substantially enhanced under tensile strain. In this paper, we have studied thermal transport of three auxetic carbon crystals: cis-C, trans-C and hin-C for short, and their strain responses by performing first-principles calculations. It is intriguing to find that their κ are much lower than those of their allotropes, and further decrease abnormally under compression. More strikingly, κ of trans-C (cis-C) anomalously increases with tensile strain up to 7% (6%) with maximum κ of almost 7 (5) times larger than the unstrained value. The abnormal strain dependent κ are attributed to the dominant role of the enhancement of phonon lifetime under stretching, which can be further explained from the unique atomic structure of the main chain of polydiacetylene in trans-C and cis-C. The weakening of phonon anharmonicity is reflected by the enhancement of root mean-square displacement values. The reported giant augmentation of κ may inspire intensive research on auxetic carbon crystals as potential materials for emerging nanoelectronic devices.},
keywords = {Auxetics, DFT, Thermal, Tubulanes},
pubstate = {published},
tppubtype = {article}
}
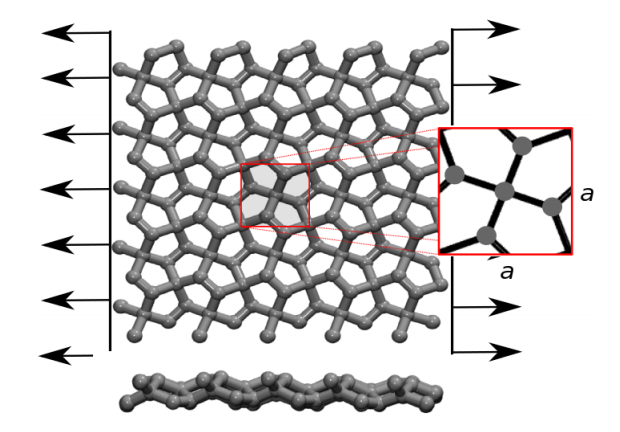
de Sousa, JM; Aguiar, AL; Girao, EC; Fonseca, Alexandre F; AG Filho, Souza; Galvao, Douglas S
Mechanical Properties and Fracture Patterns of Pentagraphene Membranes Online
2017, (preprint arXiv:1703.03789).
Resumo | Links | BibTeX | Tags: DFT, Mechanical Properties, Molecular Dynamics, pentagraphene
@online{deSousa2017,
title = {Mechanical Properties and Fracture Patterns of Pentagraphene Membranes},
author = {de Sousa, JM and Aguiar, AL and Girao, EC and Fonseca, Alexandre F and AG Filho, Souza and Galvao, Douglas S},
url = {https://arxiv.org/abs/1703.03789},
year = {2017},
date = {2017-03-10},
abstract = {Recently, a new two-dimensional carbon allotrope called pentagraphene (PG) was
proposed. PG exhibits mechanical and electronic interesting properties, including typical
band gap values of semiconducting materials. PG has a Cairo-tiling-like 2D lattice
of non coplanar pentagons and its mechanical properties have not been yet fully investigated.
In this work, we combined density functional theory (DFT) calculations and
reactive molecular dynamics (MD) simulations to investigate the mechanical properties
and fracture patterns of PG membranes under tensile strain. We show that PG
membranes can hold up to 20% of strain and that fracture occurs only after substantial
dynamical bond breaking and the formation of 7, 8 and 11 carbon rings and carbon
chains. The stress-strain behavior was observed to follow two regimes, one exhibiting linear elasticity followed by a plastic one, involving carbon atom re-hybridization with
the formation of carbon rings and chains. Our results also show that mechanically
induced structural transitions from PG to graphene is unlikely to occur, in contrast to
what was previously speculated in the literature.},
note = {preprint arXiv:1703.03789},
keywords = {DFT, Mechanical Properties, Molecular Dynamics, pentagraphene},
pubstate = {published},
tppubtype = {online}
}
proposed. PG exhibits mechanical and electronic interesting properties, including typical
band gap values of semiconducting materials. PG has a Cairo-tiling-like 2D lattice
of non coplanar pentagons and its mechanical properties have not been yet fully investigated.
In this work, we combined density functional theory (DFT) calculations and
reactive molecular dynamics (MD) simulations to investigate the mechanical properties
and fracture patterns of PG membranes under tensile strain. We show that PG
membranes can hold up to 20% of strain and that fracture occurs only after substantial
dynamical bond breaking and the formation of 7, 8 and 11 carbon rings and carbon
chains. The stress-strain behavior was observed to follow two regimes, one exhibiting linear elasticity followed by a plastic one, involving carbon atom re-hybridization with
the formation of carbon rings and chains. Our results also show that mechanically
induced structural transitions from PG to graphene is unlikely to occur, in contrast to
what was previously speculated in the literature.
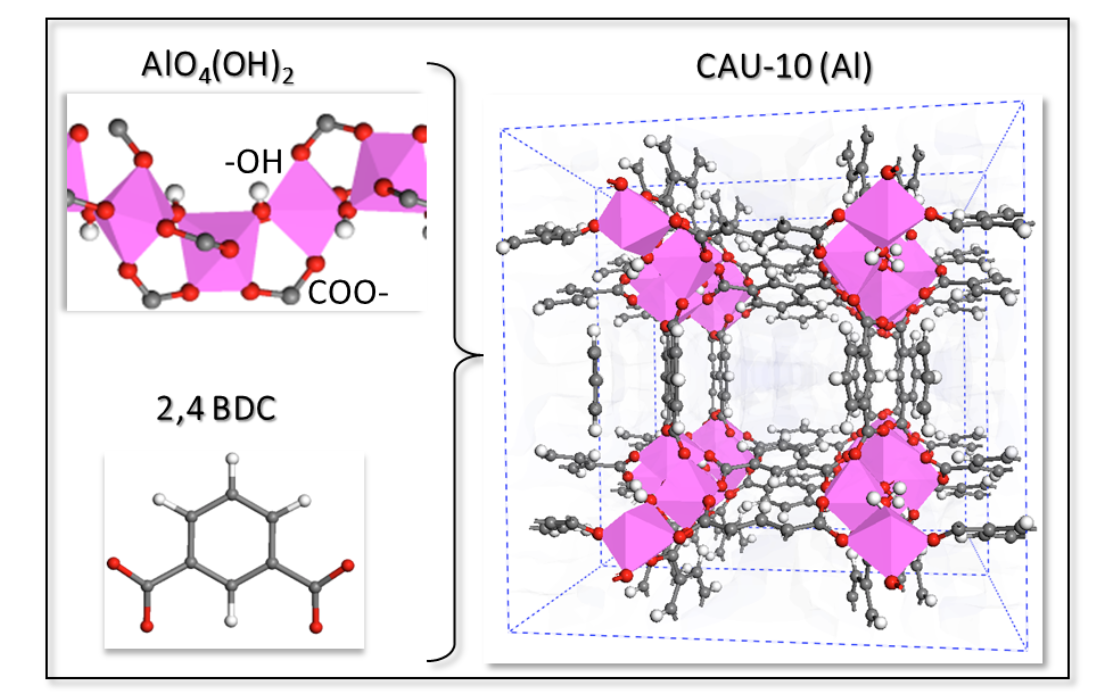
Borges, Daiane Damasceno; Maurin, Guillaume; Galvao, Douglas S
Design of Porous Metal-Organic Frameworks for Adsorption Driven Thermal Batteries Journal Article
Em: MRS Advances, vol. 2017, pp. 1-6, 2017.
Resumo | Links | BibTeX | Tags: DFT, MOFs, thermal batteries
@article{Borges2017b,
title = {Design of Porous Metal-Organic Frameworks for Adsorption Driven Thermal Batteries},
author = {Borges, Daiane Damasceno and Maurin, Guillaume and Galvao, Douglas S},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/design-of-porous-metalorganic-frameworks-for-adsorption-driven-thermal-batteries/A63B92E4D7E413D7CC047E152C7F22AF},
doi = {10.1557/adv.2017.181},
year = {2017},
date = {2017-02-15},
journal = {MRS Advances},
volume = {2017},
pages = {1-6},
abstract = {Thermal batteries based on a reversible adsorption/desorption of a working fluid (water, methanol, ammonia) rather than the conventional vapor compression is a promising alternative to exploit waste thermal energy for heat reallocation. In this context, there is an increasing interest to find novel porous solids able to adsorb a high energy density of working fluid under low relative vapor pressure condition combined with an easy ability of regeneration (desorption) at low temperature, which are the major requirements for adsorption driven heat pumps and chillers. The porous crystalline hybrid materials named Metal–Organic Frameworks (MOF) represent a great source of inspiration for sorption based-applications owing to their tunable chemical and topological features associated with a large variability of pore sizes. Recently, we have designed a new MOF named MIL-160 (MIL stands for Materials of Institut Lavoisier), isostructural to CAU-10, built from the assembly of corner sharing aluminum chains octahedra AlO4(OH)2 with the 2,5-furandicarboxylic linker substituting the pristine organic linker, 1,4-benzenedicarboxylate. This ligand replacement strategy proved to enhance both the hydrophilicity of the MOF and its amount of water adsorbed at low p/p0. This designed solid was synthesized and its chemical stability/adsorption performances verified. Here, we have extended this study by incorporating other polar heterocyclic linkers and a comparative computational study of the water adsorption performances of these novel structures has been performed. To that purpose, the cell and geometry optimizations of all hypothetical frameworks were first performed at the density functional theory level and their water adsorption isotherms were further predicted by using force-field based Grand-Canonical Monte Carlo simulations. This study reveals the ease tunable water affinity of MOF for the desired application.
},
keywords = {DFT, MOFs, thermal batteries},
pubstate = {published},
tppubtype = {article}
}
2016
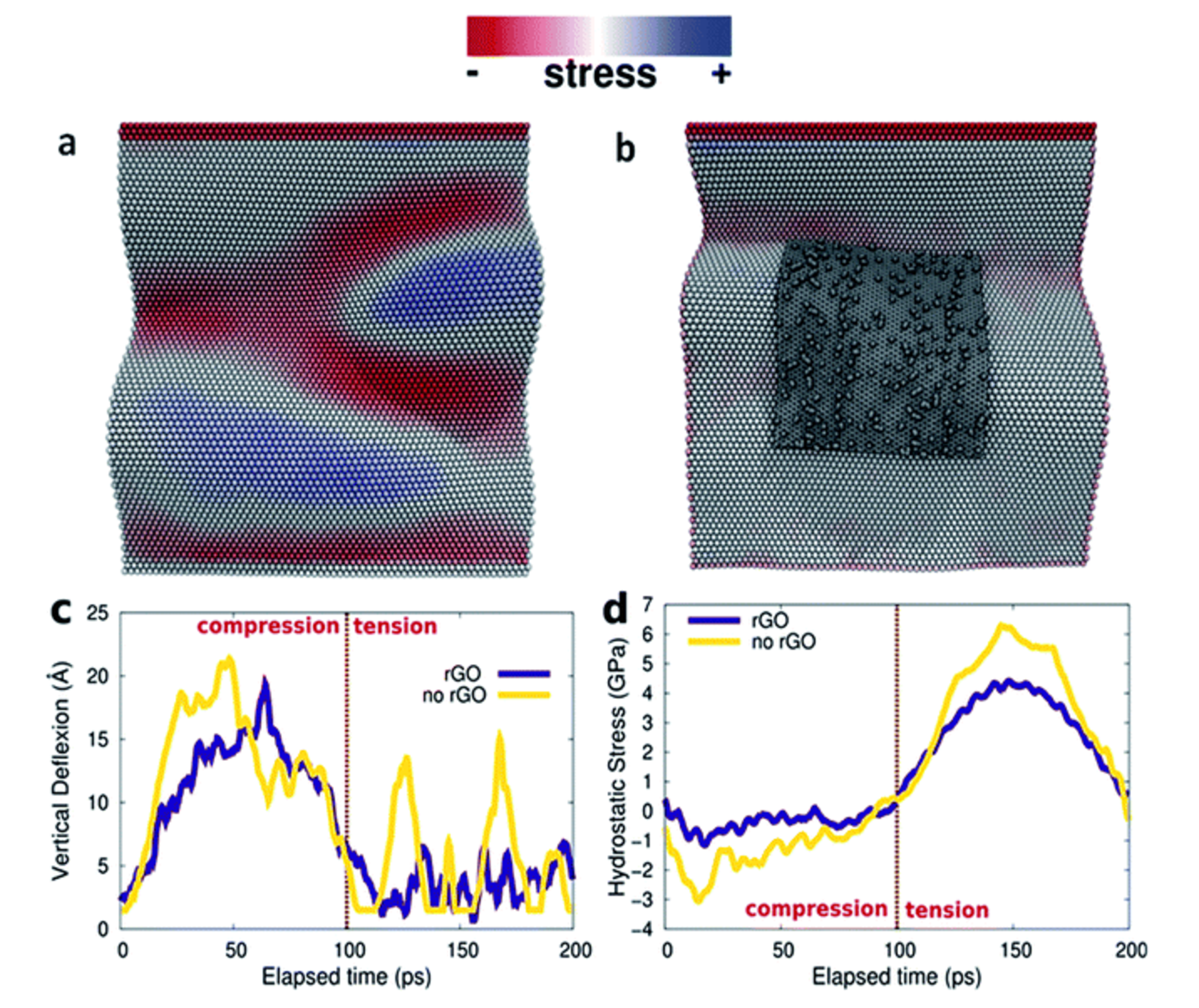
Chandra Sekhar Tiwary Sujin P Jose, Suppanat Kosolwattana
Enhanced supercapacitor performance of a 3D architecture tailored using atomically thin rGO–MoS 2 2D sheets Journal Article
Em: RSC Advances, vol. 6, pp. 93384-93393, 2016.
Resumo | Links | BibTeX | Tags: Chalcogenides, DFT, graphene oxide, Molecular Dynamics
@article{Jose2016,
title = {Enhanced supercapacitor performance of a 3D architecture tailored using atomically thin rGO–MoS 2 2D sheets},
author = {Sujin P Jose, Chandra Sekhar Tiwary, Suppanat Kosolwattana, Prasanth Raghavan, Leonardo D Machado, Chandkiram Gautam, T Prasankumar, Jarin Joyner, Sehmus Ozden, Douglas S Galvao, PM Ajayan},
url = {xlink.rsc.org/?DOI=c6ra20960b},
doi = {10.1039/C6RA20960B},
year = {2016},
date = {2016-09-19},
journal = {RSC Advances},
volume = {6},
pages = {93384-93393},
abstract = {A 3D architecture is fabricated using 2D nano-sheets of GO and MoS2 as the building blocks by a facile, one-pot chronoamperometry method to achieve a conductive additive free, binder free and scalable supercapacitor electrode. The superior electrochemical properties of the 3D PPy-rGO–MoS2 (PGMo) are due to its porous structure, thin wall, high surface area and high electrical conductivity that endow rapid transportation of electrolyte ions and electrons throughout the electrode matrix. The synergistic effect between the components in a proper ratio improves the supercapacitor performance and material stability of PGMo. The possible correlation of the structure and electrochemical performance of the 3D ternary composite is backed by a fully atomistic molecular dynamics (MD) simulation study. The high specific capacitance (387 F g−1) and impressive cycling stability (>1000 cycles) estimated for PGMo open up an opportunity to consider the 3D ternary nanostructures as cutting edge materials for energy storage solutions.
},
keywords = {Chalcogenides, DFT, graphene oxide, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Chandra Sekhar Tiwary Mohamad A Kabbani, Anirban Som
A generic approach for mechano-chemical reactions between carbon nanotubes of different functionalities Journal Article
Em: Carbon, vol. 104, pp. 196-202, 2016.
Resumo | Links | BibTeX | Tags: Carbon Nanotubes, DFT, Fracture, Mechano-chemistry, Molecular Dynamics
@article{Kabbani2016,
title = {A generic approach for mechano-chemical reactions between carbon nanotubes of different functionalities},
author = {Mohamad A Kabbani, Chandra Sekhar Tiwary, Anirban Som, KR Krishnadas, Pedro AS Autreto, Sehmus Ozden, Kunttal Keyshar, Ken Hackenberg, Alin Christian Chipara, Douglas S Galvao, Robert Vajtai, Ahmad T Kabbani, Thalappil Pradeep, Pulickel M Ajayan},
url = {www.sciencedirect.com/science/article/pii/S000862231630183X},
doi = {10.1016/j.carbon.2016.02.094},
year = {2016},
date = {2016-08-31},
journal = {Carbon},
volume = {104},
pages = {196-202},
abstract = {Abstract Here, we report similar reactions between nanotubes carrying functionalities,
namely carbon nanotubes (CNTs) with the acyl chloride/hydroxyl and amine/carboxylic
functionalities directly attached to their surfaces, resulting in the formation ofchemically
modified graphene products. The reaction is spontaneous and is facilitated by simple
grinding of the reactants. The new solid-state reactions have been confirmed using different
spectroscopic and electron microscopy techniques.},
keywords = {Carbon Nanotubes, DFT, Fracture, Mechano-chemistry, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
namely carbon nanotubes (CNTs) with the acyl chloride/hydroxyl and amine/carboxylic
functionalities directly attached to their surfaces, resulting in the formation ofchemically
modified graphene products. The reaction is spontaneous and is facilitated by simple
grinding of the reactants. The new solid-state reactions have been confirmed using different
spectroscopic and electron microscopy techniques.
Yongji Gong Bo Li, Zhili Hu
Solid–Vapor Reaction Growth of Transition‐Metal Dichalcogenide Monolayers Journal Article
Em: Angewandte Chemie, vol. 128, não 36, pp. 10814-10819, 2016.
Resumo | Links | BibTeX | Tags: Chalcogenides, cvd, DFT
@article{Li2016,
title = {Solid–Vapor Reaction Growth of Transition‐Metal Dichalcogenide Monolayers},
author = {Bo Li, Yongji Gong, Zhili Hu, Gustavo Brunetto, Yingchao Yang, Gonglan Ye, Zhuhua Zhang, Sidong Lei, Zehua Jin, Elisabeth Bianco, Xiang Zhang, Weipeng Wang, Jun Lou, Douglas S Galvão, Ming Tang, Boris I Yakobson, Robert Vajtai, Pulickel M Ajayan},
url = {onlinelibrary.wiley.com/doi/10.1002/anie.201604445/abstract},
doi = {10.1002/ange.201604445},
year = {2016},
date = {2016-08-26},
journal = {Angewandte Chemie},
volume = {128},
number = {36},
pages = {10814-10819},
abstract = {Two-dimensional (2D) layered semiconducting transition-metal dichalcogenides (TMDCs) are promising candidates for next-generation ultrathin, flexible, and transparent electronics. Chemical vapor deposition (CVD) is a promising method for their controllable, scalable synthesis but the growth mechanism is poorly understood. Herein, we present systematic studies to understand the CVD growth mechanism of monolayer MoSe2, showing reaction pathways for growth from solid and vapor precursors. Examination of metastable nanoparticles deposited on the substrate during growth shows intermediate growth stages and conversion of non-stoichiometric nanoparticles into stoichiometric 2D MoSe2 monolayers. The growth steps involve the evaporation and reduction of MoO3 solid precursors to sub-oxides and stepwise reactions with Se vapor to finally form MoSe2. The experimental results and proposed model were corroborated by ab initio Car–Parrinello molecular dynamics studies.},
keywords = {Chalcogenides, cvd, DFT},
pubstate = {published},
tppubtype = {article}
}
Rodrigo Prioli Clara M Almeida, Benjamin Fragneaud
Giant and Tunable Anisotropy of Nanoscale Friction in Graphene Journal Article
Em: Nature Scientific Reports, vol. 6, pp. 31569, 2016.
Resumo | Links | BibTeX | Tags: DFT, Graphene, Molecular Dynamics, Tribology
@article{Almeida2016,
title = {Giant and Tunable Anisotropy of Nanoscale Friction in Graphene},
author = {Clara M Almeida, Rodrigo Prioli, Benjamin Fragneaud, Luiz Gustavo Cançado, Ricardo Paupitz, Douglas S Galvão, Marcelo De Cicco, Marcos G Menezes, Carlos A Achete, Rodrigo B Capaz},
url = {http://www-nature-com.ez88.periodicos.capes.gov.br/articles/srep31569},
doi = {10.1038/srep31569},
year = {2016},
date = {2016-07-18},
journal = {Nature Scientific Reports},
volume = {6},
pages = {31569},
abstract = {The nanoscale friction between an atomic force microscopy tip and graphene is investigated using friction force microscopy (FFM). During the tip movement, friction forces are observed to increase and then saturate in a highly anisotropic manner. As a result, the friction forces in graphene are highly dependent on the scanning direction: under some conditions, the energy dissipated along the armchair direction can be 80% higher than along the zigzag direction. In comparison, for highly-oriented pyrolitic graphite (HOPG), the friction anisotropy between armchair and zigzag directions is only 15%. This giant friction anisotropy in graphene results from anisotropies in the amplitudes of flexural deformations of the graphene sheet driven by the tip movement, not present in HOPG. The effect can be seen as a novel manifestation of the classical phenomenon of Euler buckling at the nanoscale, which provides the non-linear ingredients that amplify friction anisotropy. Simulations based on a novel version of the 2D Tomlinson model (modified to include the effects of flexural deformations), as well as fully atomistic molecular dynamics simulations and first-principles density-functional theory (DFT) calculations, are able to reproduce and explain the experimental observations.
},
keywords = {DFT, Graphene, Molecular Dynamics, Tribology},
pubstate = {published},
tppubtype = {article}
}
2014
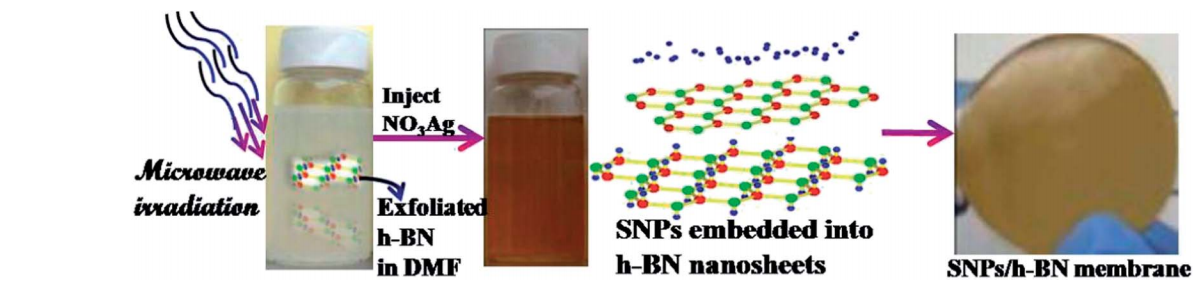
Gao, Guanhui; Mathkar, Akshay; Martins, Eric Perim; Galvao, Douglas S; Gao, Duyang; da Silva Autreto, Pedro Alves; Sun, Chengjun; Cai, Lintao; Ajayan, Pulickel M
Designing nanoscaled hybrids from atomic layered boron nitride with silver nanoparticle deposition Journal Article
Em: Journal of Materials Chemistry A, vol. 2, não 9, pp. 3148–3154, 2014.
Resumo | Links | BibTeX | Tags: DFT, nano particles
@article{gao2014designing,
title = {Designing nanoscaled hybrids from atomic layered boron nitride with silver nanoparticle deposition},
author = {Gao, Guanhui and Mathkar, Akshay and Martins, Eric Perim and Galvao, Douglas S and Gao, Duyang and da Silva Autreto, Pedro Alves and Sun, Chengjun and Cai, Lintao and Ajayan, Pulickel M},
url = {http://pubs.rsc.org/en/Content/ArticleLanding/2014/TA/c3ta12892j#!divAbstract},
year = {2014},
date = {2014-01-01},
journal = {Journal of Materials Chemistry A},
volume = {2},
number = {9},
pages = {3148--3154},
publisher = {Royal Society of Chemistry},
abstract = {We have developed a microwave assisted one-pot approach to fabricate a novel hybrid nano-composite composed of two-dimensional chemically exfoliated layered hexagonal boron nitride (h-BN) and embedded silver nanoparticles (SNP). Atomic layered h-BN exfoliated using chemical liquid showed strong in-plane bonding and weak van der Waals interplanar interactions, which is utilized for chemically interfacing SNP, indicating their ability to act as excellent nano-scaffolds. The SNP/h-BN optical response, in particular band gap, is strongly dependent on the concentration of the metallic particles. In order to gain further insight into this behavior we have also carried out ab initio density functional theory (DFT) calculations on modeled structures, demonstrating that the bandgap value of SNP/h-BN hybrids could be significantly altered by a small percentage of OH− groups located at dangling B and N atoms. Our results showed that these novel SNP/h-BN nanohybrid structures exhibited excellent thermal stability and they are expected to be applied as devices for thermal oxidation-resistant surface enhanced Raman spectroscopy (SERS). The SNP/h-BN membrane showed remarkable antibacterial activity, suggesting their potential use in water disinfection and food packaging.
},
keywords = {DFT, nano particles},
pubstate = {published},
tppubtype = {article}
}
Perim, E; Paupitz, R; Botari, T; Galvao, DS
One-dimensional silicon and germanium nanostructures with no carbon analogues Journal Article
Em: Physical Chemistry Chemical Physics, vol. 16, não 44, pp. 24570–24574, 2014.
Resumo | Links | BibTeX | Tags: DFT, Germanium, Nanotubes, Silicon
@article{perim2014one,
title = {One-dimensional silicon and germanium nanostructures with no carbon analogues},
author = {Perim, E and Paupitz, R and Botari, T and Galvao, DS},
url = {http://pubs.rsc.org/en/content/articlehtml/2014/cp/c4cp03708a},
year = {2014},
date = {2014-01-01},
journal = {Physical Chemistry Chemical Physics},
volume = {16},
number = {44},
pages = {24570--24574},
publisher = {Royal Society of Chemistry},
abstract = {In this work we report new silicon and germanium tubular nanostructures with no corresponding stable carbon analogues. The electronic and mechanical properties of these new tubes were investigated through ab initio methods. Our results show that these structures have lower energy than their corresponding nanoribbon structures and are stable up to high temperatures (500 and 1000 K, for silicon and germanium tubes, respectively). Both tubes are semiconducting with small indirect band gaps, which can be significantly altered by both compressive and tensile strains. Large bandgap variations of almost 50% were observed for strain rates as small as 3%, suggesting their possible applications in sensor devices. They also present high Young's modulus values (0.25 and 0.15 TPa, respectively). TEM images were simulated to help in the identification of these new structures.},
keywords = {DFT, Germanium, Nanotubes, Silicon},
pubstate = {published},
tppubtype = {article}
}
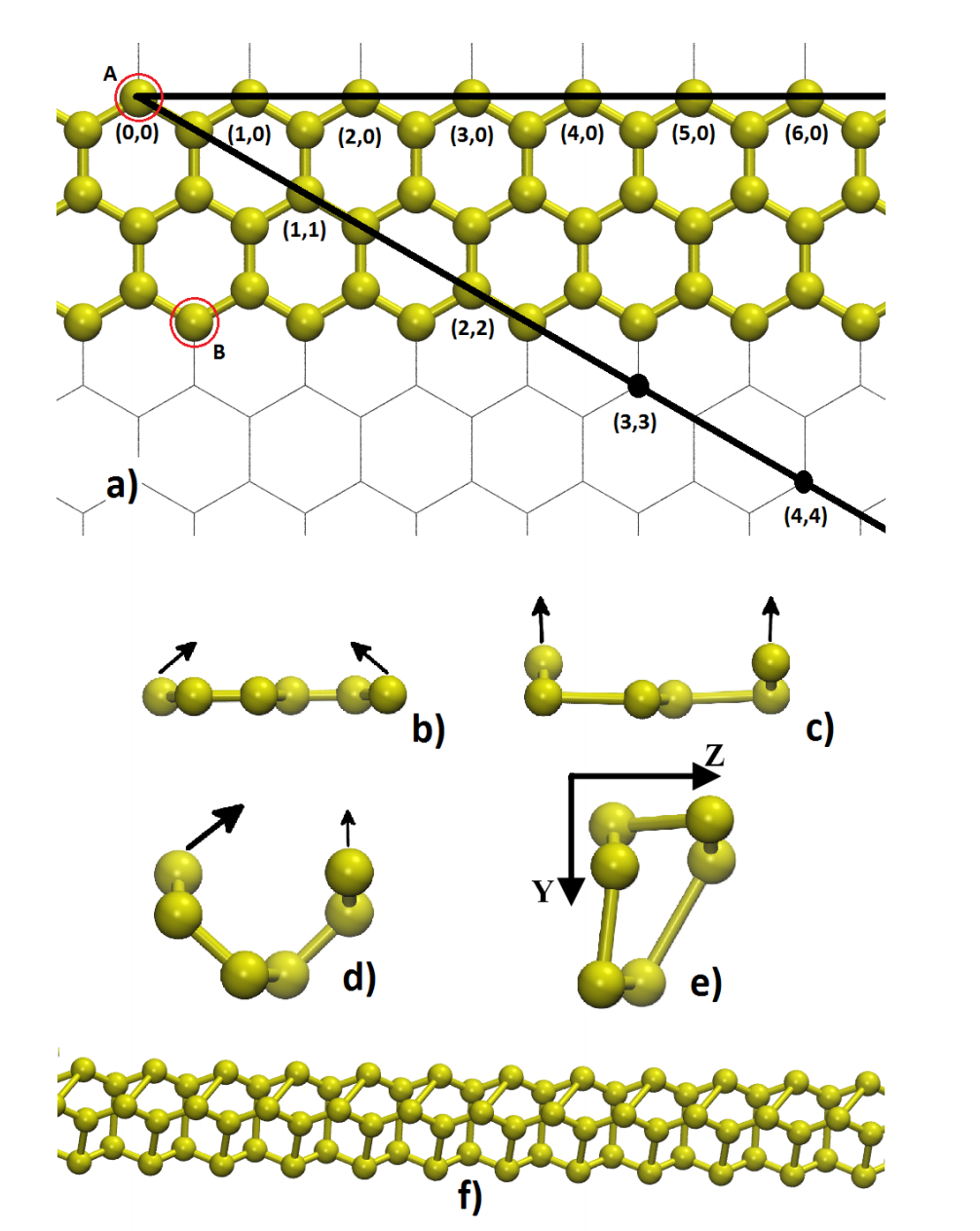
Perim, Eric; Paupitz, Ricardo; Botari, Tiago; Galvao, Douglas S
Novel Semiconducting Silicon and Germanium Nanotubes Journal Article
Em: arXiv preprint arXiv:1403.2061, 2014.
Resumo | Links | BibTeX | Tags: DFT, Germanium, Nanotubes, Silicon
@article{perim2014novel,
title = {Novel Semiconducting Silicon and Germanium Nanotubes},
author = {Perim, Eric and Paupitz, Ricardo and Botari, Tiago and Galvao, Douglas S},
url = {http://arxiv.org/abs/1403.2061},
year = {2014},
date = {2014-01-01},
journal = {arXiv preprint arXiv:1403.2061},
abstract = {In this work we report new silicon and germanium nanotube structures, with no corresponding
stable carbon analogues and which cannot be described by integer chiral indices. The electronic
and mechanical properties of these new tubes were investigated through ab initio methods. Our
results show that the structures are stable up to high temperatures (500 and 1000 K, for silicon and
germanium tubes, respectively). Both tubes are semiconducting with small indirect band gaps,
which can be significantly altered by both compressive and tensile strains. They also present high
Young modulus values (0.25 and 0.15 TPa, respectively).},
keywords = {DFT, Germanium, Nanotubes, Silicon},
pubstate = {published},
tppubtype = {article}
}
stable carbon analogues and which cannot be described by integer chiral indices. The electronic
and mechanical properties of these new tubes were investigated through ab initio methods. Our
results show that the structures are stable up to high temperatures (500 and 1000 K, for silicon and
germanium tubes, respectively). Both tubes are semiconducting with small indirect band gaps,
which can be significantly altered by both compressive and tensile strains. They also present high
Young modulus values (0.25 and 0.15 TPa, respectively).
2013
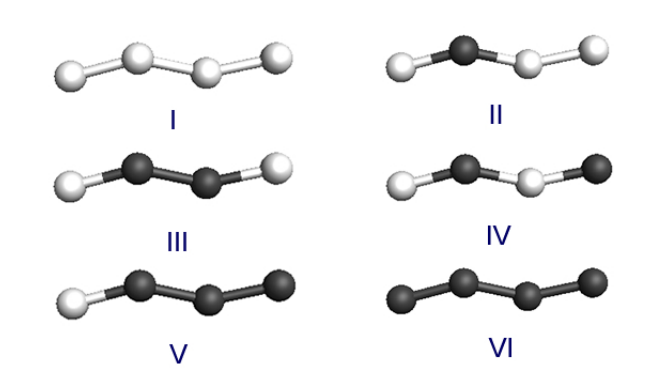
Autreto, Pedro Alves da Silva; Galvao, Douglas S; Artacho, Emilio
Species Fractionation in Atomic Chains from Mechanically Stretched Alloys Journal Article
Em: arXiv preprint arXiv:1312.1285, 2013.
Resumo | Links | BibTeX | Tags: Atomic Chains, DFT, Mech, Mechanical Properties, Metallic Nanowires
@article{autreto2013species,
title = {Species Fractionation in Atomic Chains from Mechanically Stretched Alloys},
author = {Autreto, Pedro Alves da Silva and Galvao, Douglas S and Artacho, Emilio},
url = {http://arxiv.org/abs/1312.1285},
year = {2013},
date = {2013-01-01},
journal = {arXiv preprint arXiv:1312.1285},
abstract = {Bettini et al. [Nature Nanotech 1, 182 (2006)] reported the first experimental realization of linear
atomic chains (LACs) composed of different atoms (Au and Ag). Different contents of Au and Ag
were observed in the chains from what found in the bulk alloys, which rises the question of what is the
wire composition if in equilibrium with a bulk alloy. In this work we address the thermodynamic
driving force for species fractionation in LACs under tension, and we present density-functional
theory results for Ag-Au chain alloys. A pronounced stabilization of wires with an alternating
Ag-Au sequence is observed, which could be behind the experimentally observed Au enrichment in
LACs from alloys of high Ag content.},
keywords = {Atomic Chains, DFT, Mech, Mechanical Properties, Metallic Nanowires},
pubstate = {published},
tppubtype = {article}
}
atomic chains (LACs) composed of different atoms (Au and Ag). Different contents of Au and Ag
were observed in the chains from what found in the bulk alloys, which rises the question of what is the
wire composition if in equilibrium with a bulk alloy. In this work we address the thermodynamic
driving force for species fractionation in LACs under tension, and we present density-functional
theory results for Ag-Au chain alloys. A pronounced stabilization of wires with an alternating
Ag-Au sequence is observed, which could be behind the experimentally observed Au enrichment in
LACs from alloys of high Ag content.
2012
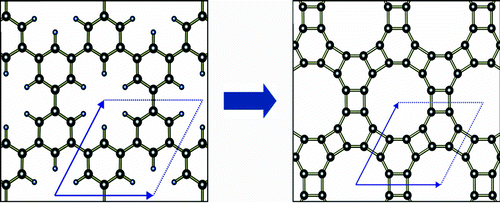
Brunetto, Gustavo; Autreto, PAS; Machado, Leonardo Dantas; Santos, BI; dos Santos, Ricardo PB; Galvao, Douglas S
Nonzero gap two-dimensional carbon allotrope from porous graphene Journal Article
Em: The Journal of Physical Chemistry C, vol. 116, não 23, pp. 12810–12813, 2012.
Resumo | Links | BibTeX | Tags: BPC, DFT, Graphene, Porous Graphene
@article{brunetto2012nonzero,
title = {Nonzero gap two-dimensional carbon allotrope from porous graphene},
author = {Brunetto, Gustavo and Autreto, PAS and Machado, Leonardo Dantas and Santos, BI and dos Santos, Ricardo PB and Galvao, Douglas S},
url = {http://pubs.acs.org/doi/abs/10.1021/jp211300n},
year = {2012},
date = {2012-01-01},
journal = {The Journal of Physical Chemistry C},
volume = {116},
number = {23},
pages = {12810--12813},
publisher = {American Chemical Society},
abstract = {Graphene is considered one of the most promising materials for future electronics. However, in its pristine form, graphene is a gapless material, which imposes limitations to its use in some electronic applications. To solve this problem, many approaches have been tried, such as physical and chemical functionalizations. These processes compromise some of the desirable graphene properties. In this work, based on ab initio quantum molecular dynamics, we showed that a two-dimensional carbon allotrope, named biphenylene carbon (BPC), can be obtained from selective dehydrogenation of porous graphene. BPC presents a nonzero bandgap and well-delocalized frontier orbitals. Synthetic routes to BPC are also addressed.},
keywords = {BPC, DFT, Graphene, Porous Graphene},
pubstate = {published},
tppubtype = {article}
}
2011
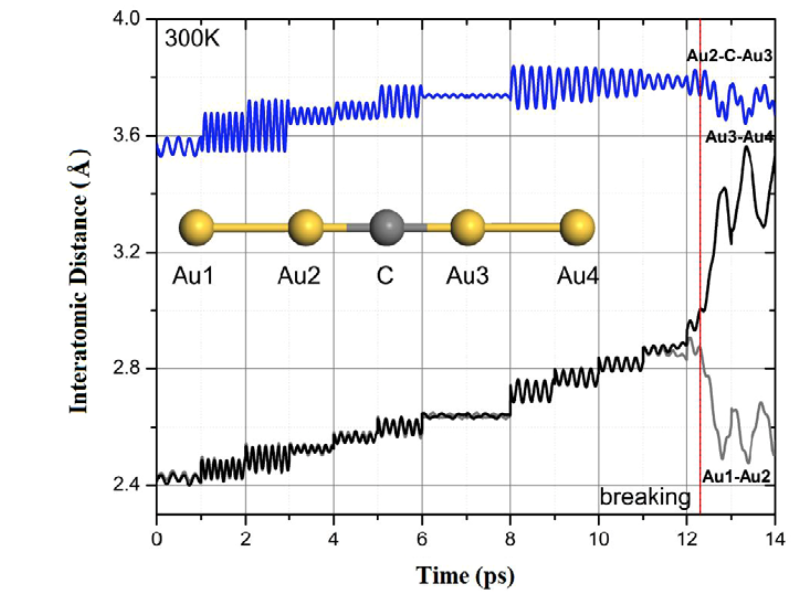
PAS Autreto MJ Lagos, SB Legoas
Temperature effects on the occurrence of long interatomic distances in atomic chains formed from stretched gold nanowires Journal Article
Em: Nanotechnology, vol. 22, não 9, pp. 095705, 2011.
Resumo | Links | BibTeX | Tags: DFT, Gold, Metallic Nanowires, TEM
@article{Lagos2011,
title = {Temperature effects on the occurrence of long interatomic distances in atomic chains formed from stretched gold nanowires},
author = {MJ Lagos, PAS Autreto, SB Legoas, F Sato, V Rodrigues, DS Galvao, D Ugarte},
url = {http://iopscience.iop.org/0957-4484/22/9/095705},
year = {2011},
date = {2011-03-04},
journal = {Nanotechnology},
volume = {22},
number = {9},
pages = {095705},
abstract = {The origin of long interatomic distances in suspended gold atomic chains formed from stretched nanowires remains the object of debate despite the large amount of theoretical and experimental work. Here, we report new atomic resolution electron microscopy observations acquired at room and liquid-nitrogen temperatures and theoretical results from ab initio quantum molecular dynamics on chain formation and stability. These new data are suggestive that the long distances are due to contamination by carbon atoms originating from the decomposition of adsorbed hydrocarbon molecules.
},
keywords = {DFT, Gold, Metallic Nanowires, TEM},
pubstate = {published},
tppubtype = {article}
}

Autreto, PAS; Lagos, MJ; Sato, F; Bettini, J; Rocha, AR; Rodrigues, V; Ugarte, D; Galvao, DS
Intrinsic Stability of the Smallest Possible Silver Nanotube Journal Article
Em: Physical Review Letters, vol. 106, não 6, pp. 065501, 2011.
Resumo | Links | BibTeX | Tags: DFT, Mechanical Properties, Metallic Nanowires, New Structures, top20
@article{autreto2011intrinsic,
title = {Intrinsic Stability of the Smallest Possible Silver Nanotube},
author = {Autreto, PAS and Lagos, MJ and Sato, F and Bettini, J and Rocha, AR and Rodrigues, V and Ugarte, D and Galvao, DS},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.106.065501},
year = {2011},
date = {2011-01-01},
journal = {Physical Review Letters},
volume = {106},
number = {6},
pages = {065501},
publisher = {American Physical Society},
abstract = {Recently, Lagos et al. [Nature Nanotech. 4, 149 (2009)] reported the discovery of the smallest possible Ag nanotube with a square cross section. Ab initio density functional theory calculations strongly support that the stability of these hollow structures is structurally intrinsic and not the result of contamination by light atoms. We also report the first experimental observation of the theoretically predicted corrugation of the hollow structure. Quantum conductance calculations predict a unique signature of 3.6G0 for this new family of nanotubes.},
keywords = {DFT, Mechanical Properties, Metallic Nanowires, New Structures, top20},
pubstate = {published},
tppubtype = {article}
}
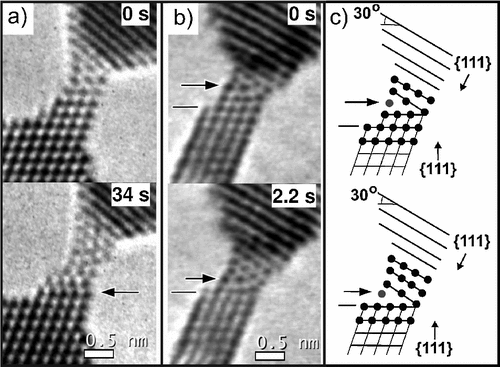
Lagos, Maureen J; Sato, Fernando; Galvao, Douglas S; Ugarte, Daniel
Mechanical deformation of nanoscale metal rods: when size and shape matter Journal Article
Em: Physical Review Letters, vol. 106, não 5, pp. 055501, 2011.
Resumo | Links | BibTeX | Tags: Defects, DFT, Mechanical Properties, Metallic Nanowires
@article{lagos2011mechanical,
title = {Mechanical deformation of nanoscale metal rods: when size and shape matter},
author = {Lagos, Maureen J and Sato, Fernando and Galvao, Douglas S and Ugarte, Daniel},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.106.055501},
year = {2011},
date = {2011-01-01},
journal = {Physical Review Letters},
volume = {106},
number = {5},
pages = {055501},
publisher = {American Physical Society},
abstract = {Face centered cubic metals deform mainly by propagating partial dislocations generating planar fault ribbons. How do metals deform if the size is smaller than the fault ribbons? We studied the elongation of Au and Pt nanorods by in situ electron microscopy and ab initio calculations. Planar fault activation barriers are so low that, for each temperature, a minimal rod size is required to become active for releasing elastic energy. Surface effects dominate deformation energetics; system size and shape determine the preferred fault gliding directions which induce different tensile and compressive behavior.
},
keywords = {Defects, DFT, Mechanical Properties, Metallic Nanowires},
pubstate = {published},
tppubtype = {article}
}
Coutinho, Samir S; Azevedo, David L; Galvao, Douglas S
Tuning Electronic and Structural Properties of Triple Layers of Intercalated Graphene and Hexagonal Boron Nitride: An Ab-initio Study. Journal Article
Em: MRS Proceedings, vol. 1307, pp. mrsf10–1307, 2011.
Resumo | Links | BibTeX | Tags: BN, DFT, Graphene, Heterostructures
@article{coutinho2011tuning,
title = {Tuning Electronic and Structural Properties of Triple Layers of Intercalated Graphene and Hexagonal Boron Nitride: An Ab-initio Study.},
author = {Coutinho, Samir S and Azevedo, David L and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8330317&fileId=S1946427411003642},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1307},
pages = {mrsf10--1307},
publisher = {Cambridge University Press},
abstract = {Recently, several experiments and theoretical studies demonstrated the possibility of tuning or modulating band gap values of nanostructures composed of bi-layer graphene, bi-layer hexagonal boron-nitride (BN) and hetero-layer combinations. These triple layers systems present several possibilities of stacking. In this work we report an ab initio (within the formalism of density functional theory (DFT)) study of structural and electronic properties of some of these stacked configurations. We observe that an applied external electric field can alter the electronic and structural properties of these systems. With the same value of the applied electric field the band gap values can be increased or decreased, depending on the layer stacking sequences. Strong geometrical deformations were observed. These results show that the application of an external electric field perpendicular to the stacked layers can effectively be used to modulate their inter-layer distances and/or their band gap values.},
keywords = {BN, DFT, Graphene, Heterostructures},
pubstate = {published},
tppubtype = {article}
}
2010
Sato, F; Legoas, SB; Otero, R; Hummelink, F; Thostrup, P; Lægsgaard, E; Stensgaard, I; Besenbacher, F; Galvao, DS
Adsorption configuration effects on the surface diffusion of large organic molecules: The case of Violet Lander Journal Article
Em: The Journal of chemical physics, vol. 133, não 22, pp. 224702, 2010.
Resumo | Links | BibTeX | Tags: DFT, Diffusion, Molecular Electronics, STM, Violet Lander
@article{sato2010adsorption,
title = {Adsorption configuration effects on the surface diffusion of large organic molecules: The case of Violet Lander},
author = {Sato, F and Legoas, SB and Otero, R and Hummelink, F and Thostrup, P and Lægsgaard, E and Stensgaard, I and Besenbacher, F and Galvao, DS},
url = {http://scitation.aip.org/content/aip/journal/jcp/133/22/10.1063/1.3512623},
year = {2010},
date = {2010-01-01},
journal = {The Journal of chemical physics},
volume = {133},
number = {22},
pages = {224702},
publisher = {AIP Publishing},
abstract = {Violet Lander (C108H104) is a large organic molecule that when deposited on Cu(110) surface exhibits lock-and-key like behavior [Otero et al., Nature Mater. 3, 779 (2004)]. In this work, we report a detailed fully atomistic molecular mechanics and molecular dynamics study of this phenomenon. Our results show that it has its physical basis on the interplay of the molecular hydrogens and the Cu(110) atomic spacing, which is a direct consequence of the matching between molecule and surface dimensions. This information could be used to find new molecules capable of displaying lock-and-key behavior with new potential applications in nanotechnology.},
keywords = {DFT, Diffusion, Molecular Electronics, STM, Violet Lander},
pubstate = {published},
tppubtype = {article}
}
Autreto, PAS; Legoas, SB; Flores, MZS; Galvao, DS
Carbon nanotube with square cross-section: An ab initio investigation Journal Article
Em: The Journal of chemical physics, vol. 133, não 12, pp. 124513, 2010.
Resumo | Links | BibTeX | Tags: Carbon Nanotubes, DFT, New Structures, square tubes
@article{autreto2010carbon,
title = {Carbon nanotube with square cross-section: An ab initio investigation},
author = {Autreto, PAS and Legoas, SB and Flores, MZS and Galvao, DS},
url = {http://scitation.aip.org/content/aip/journal/jcp/133/12/10.1063/1.3483237},
year = {2010},
date = {2010-01-01},
journal = {The Journal of chemical physics},
volume = {133},
number = {12},
pages = {124513},
publisher = {AIP Publishing},
abstract = {Recently, Lagos et al. [Nat. Nanotechnol.4, 149 (2009)] reported the discovery of the smallest possible silver square cross-section nanotube. A natural question is whether similar carbon nanotubes can exist. In this work we report ab initio results for the structural, stability, and electronic properties for such hypothetical structures. Our results show that stable (or at least metastable) structures are possible with metallic properties. They also show that these structures can be obtained by a direct interconversion from SWNT(2,2). Large finite cubanelike oligomers, topologically related to these new tubes, were also investigated.
},
keywords = {Carbon Nanotubes, DFT, New Structures, square tubes},
pubstate = {published},
tppubtype = {article}
}
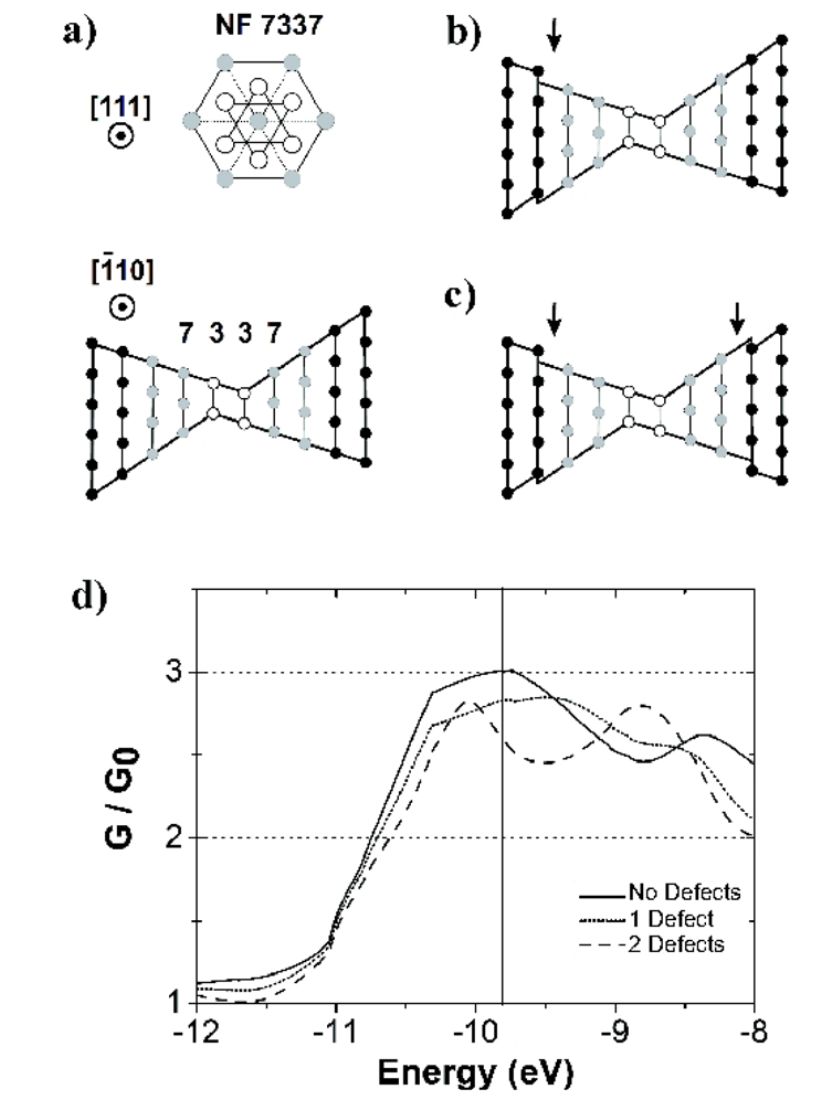
Lagos, MJ; Sato, F; Autreto, PAS; Galvao, DS; Rodrigues, V; Ugarte, D
Temperature effects on the atomic arrangement and conductance of atomic-size gold nanowires generated by mechanical stretching Journal Article
Em: Nanotechnology, vol. 21, não 48, pp. 485702, 2010.
Resumo | Links | BibTeX | Tags: DFT, Mechanical Properties, Metallic Nanowires, Quantum Transport, TEM
@article{lagos2010temperature,
title = {Temperature effects on the atomic arrangement and conductance of atomic-size gold nanowires generated by mechanical stretching},
author = {Lagos, MJ and Sato, F and Autreto, PAS and Galvao, DS and Rodrigues, V and Ugarte, D},
url = {http://iopscience.iop.org/0957-4484/21/48/485702},
year = {2010},
date = {2010-01-01},
journal = {Nanotechnology},
volume = {21},
number = {48},
pages = {485702},
publisher = {IOP Publishing},
abstract = {We have studied the changes induced by thermal effects in the structural and transport response of Au nanowires generated by mechanical elongation. We have used time-resolved atomic resolution transmission electron microscopy imaging and quantum conductance measurement using a mechanically controllable break junction. Our results showed remarkable differences in the NW evolution for experiments realized at 150 and 300 K, which modifies drastically the conductance response during elongation. Molecular dynamics and electronic transport calculations were used to consistently correlate the observed structural and conductance behavior. These results emphasize that it is essential to take into account the precise atomic arrangement of nanocontacts generated by mechanical stretching to understand electrical transport properties. Also, our study shows that much care must be taken when comparing results obtained in different experimental conditions, mainly different temperatures.
},
keywords = {DFT, Mechanical Properties, Metallic Nanowires, Quantum Transport, TEM},
pubstate = {published},
tppubtype = {article}
}
2008

E. W. S.; Freire Caetano, V. N. ; dos Santos
Mobius and twisted graphene nanoribbons: Stability, geometry, and electronic properties Journal Article
Em: THE JOURNAL OF CHEMICAL PHYSICS, vol. 128, pp. 164719, 2008.
Resumo | Links | BibTeX | Tags: DFT, Graphene, Mobis, NanoRibbons, Structure
@article{Caetano2008,
title = {Mobius and twisted graphene nanoribbons: Stability, geometry, and electronic properties},
author = {Caetano, E. W. S.; Freire, V. N.; dos Santos, S. G.; Galvao, D. S.,and Sato, F.},
url = {http://scitation.aip.org/content/aip/journal/jcp/128/16/10.1063/1.2908739},
year = {2008},
date = {2008-04-29},
journal = {THE JOURNAL OF CHEMICAL PHYSICS},
volume = {128},
pages = {164719},
abstract = {Results of classical force field geometry optimizations for twisted graphenenanoribbons with a number of twists Nt varying from 0 to 7 (the case Nt=1 corresponds to a half-twist Möbius nanoribbon) are presented in this work. Their structural stability was investigated using the Brenner reactive force field. The best classical molecular geometries were used as input for semiempirical calculations, from which the electronic properties (energy levels, HOMO, LUMO orbitals) were computed for each structure. CI wavefunctions were also calculated in the complete active space framework taking into account eigenstates from HOMO−4 to LUMO+4, as well as the oscillator strengths corresponding to the first optical transitions in the UV-VIS range. The lowest energy molecules were found less symmetric than initial configurations, and the HOMO-LUMO energy gaps are larger than the value found for the nanographene used to build them due to electronic localization effects created by the twisting. A high number of twists leads to a sharp increase of the HOMO→LUMO transition energy. We suggest that some twisted nanoribbons could form crystals stabilized by dipolar interactions.},
keywords = {DFT, Graphene, Mobis, NanoRibbons, Structure},
pubstate = {published},
tppubtype = {article}
}
Rurali, R; Cartoixa, X; Galvao, DS
Large electromechanical response in silicon nanowires predicted from first-principles electronic structure calculations Journal Article
Em: Physical Review B, vol. 77, não 7, pp. 073403, 2008.
Resumo | Links | BibTeX | Tags: DFT, Eletroactuation, Nanowires, Silicon
@article{rurali2008large,
title = {Large electromechanical response in silicon nanowires predicted from first-principles electronic structure calculations},
author = {Rurali, R and Cartoixa, X and Galvao, DS},
url = {http://journals.aps.org/prb/abstract/10.1103/PhysRevB.77.073403},
year = {2008},
date = {2008-01-01},
journal = {Physical Review B},
volume = {77},
number = {7},
pages = {073403},
publisher = {American Physical Society},
abstract = {We study by means of first-principles electronic structure calculations the electromechanical response, i.e., the structural modifications upon charge injection, of ⟨100⟩ silicon nanowires. We show that, at variance with sp2 carbon nanostructures, the response is remarkably linear, discriminates between injected charge of different signs, and is up to one order of magnitude larger than in carbon nanotubes.
},
keywords = {DFT, Eletroactuation, Nanowires, Silicon},
pubstate = {published},
tppubtype = {article}
}
Konstantinova, Elena; Camilo Jr, Alexandre; Barone, Paulo MVB; Dantas, Socrates O; Galvao, Douglas S
Some electronic properties of saturated and unsaturated cubane oligomers using DFT-based calculations Journal Article
Em: Journal of Molecular Structure: THEOCHEM, vol. 868, não 1, pp. 37–41, 2008.
Resumo | Links | BibTeX | Tags: Cubanes, DFT, Polymer
@article{konstantinova2008some,
title = {Some electronic properties of saturated and unsaturated cubane oligomers using DFT-based calculations},
author = {Konstantinova, Elena and Camilo Jr, Alexandre and Barone, Paulo MVB and Dantas, Socrates O and Galvao, Douglas S},
url = {http://www.sciencedirect.com/science/article/pii/S016612800800448X},
year = {2008},
date = {2008-01-01},
journal = {Journal of Molecular Structure: THEOCHEM},
volume = {868},
number = {1},
pages = {37--41},
publisher = {Elsevier},
abstract = {Cubanes and cubane-based molecular structures attract considerable interest as structural units which represent a new class of materials with remarkable properties. These structures are potentially useful for a variety of industrial applications and, for this reason, deserve detailed study. One of the options is to use cubane-based structures to synthesize a new class of conducting polymers with small energy band gap. In the present work we use the DFT-based methods to perform geometrical optimization and obtain some electronic properties for cubane, cubatriene, saturated and unsaturated oligomers containing different number of cubane and cubatriene building units. Our results indicate that the energy difference between the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) manifests a small decrease with the growing units number for saturated or unsaturated oligomers. This energy difference is strongly dependent on the presence of hydrogen atoms and is greater for unsaturated structures.},
keywords = {Cubanes, DFT, Polymer},
pubstate = {published},
tppubtype = {article}
}
2007
Rodrigues, Varlei; Sato, Fernando; Galvao, Douglas S; Ugarte, Daniel
Is Small Perfect? Size Limit to Defect Formation in Pyramidal Pt Nanocontacts Journal Article
Em: arXiv preprint arXiv:0707.4187, 2007.
Resumo | Links | BibTeX | Tags: Defects, DFT, Metallic Nanowires, Structure, TEM
@article{rodrigues2007small,
title = {Is Small Perfect? Size Limit to Defect Formation in Pyramidal Pt Nanocontacts},
author = {Rodrigues, Varlei and Sato, Fernando and Galvao, Douglas S and Ugarte, Daniel},
url = {http://xxx.tau.ac.il/abs/0707.4187},
year = {2007},
date = {2007-01-01},
journal = {arXiv preprint arXiv:0707.4187},
abstract = {We report high resolution transmission electron microscopy and ab initio calculation results for the defect formation in Pt nanocontacts (NCs). Our results show that there is a size limit to the existence of twins (extended structural defects). Defects are always present but blocked away from the tip axes. The twins may act as scattering plane, influencing contact electron transmission for Pt NC at room temperature and Ag/Au NC at low temperature.},
keywords = {Defects, DFT, Metallic Nanowires, Structure, TEM},
pubstate = {published},
tppubtype = {article}
}
Rodrigues, V; Sato, F; Galvao, DS; Ugarte, D
Size limit of defect formation in pyramidal Pt nanocontacts Journal Article
Em: Physical Review Letters, vol. 99, não 25, pp. 255501, 2007.
Resumo | Links | BibTeX | Tags: DFT, Metallic Nanowires, Platinum, Structure, TEM, top20
@article{rodrigues2007size,
title = {Size limit of defect formation in pyramidal Pt nanocontacts},
author = {Rodrigues, V and Sato, F and Galvao, DS and Ugarte, D},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.99.255501},
year = {2007},
date = {2007-01-01},
journal = {Physical Review Letters},
volume = {99},
number = {25},
pages = {255501},
publisher = {American Physical Society},
abstract = {We report high resolution transmission electron microscopy and ab initio calculation results for defect formation in sharp pyramidal Pt nanocontacts. Our results show that there is a size limit to the existence of twins (extended structural defects). These defects are always present but blocked away from the tip axes. They may act as scattering planes, influencing the electron conductance for Pt nanocontacts at room temperature and Ag/Au nanocontacts at low temperature (<150 K).},
keywords = {DFT, Metallic Nanowires, Platinum, Structure, TEM, top20},
pubstate = {published},
tppubtype = {article}
}
2006
Troche, KS; Coluci, VR; Rurali, R; Galvao, DS
Doping of zigzag carbon nanotubes through the encapsulation of small fullerenes Journal Article
Em: arXiv preprint cond-mat/0607197, 2006.
Resumo | Links | BibTeX | Tags: CNT encapsulation, DFT, Molecular Dynamics
@article{troche2006doping,
title = {Doping of zigzag carbon nanotubes through the encapsulation of small fullerenes},
author = {Troche, KS and Coluci, VR and Rurali, R and Galvao, DS},
url = {http://arxiv.org/abs/cond-mat/0607197},
year = {2006},
date = {2006-01-01},
journal = {arXiv preprint cond-mat/0607197},
abstract = {In this work we investigated the encapsulation of C20 and C30 fullerenes into semiconducting carbon nanotubes to study the possibility of bandgap engineering in such systems. Classical molecular dynamics simulations coupled to tight-binding calculations were used to determine the conformational and electronic properties of carbon nanotube supercells containing up to 12 fullerenes. We have observed that C20 fullerenes behave similarly to a p-type dopant while C30 ones work as n-type ones. For larger diameter nanotubes, where fullerene patterns start to differ from the linear arrangements (peapods), the doping features are preserved for both fullerenes, but local disorder plays an important role and significantly alters the electronic structure. The combined incorporation of both fullerene types (hybrid encapsulation) into the same nanotube leads to a behavior similar to that found in electronic junctions in Silicon-based electronic devices. These aspects can be exploited in the design of nanoelectronic devices using semiconducting carbon nanotubes.},
keywords = {CNT encapsulation, DFT, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Coluci, Vitor R; Galvao, Douglas S; Jorio, A
Geometric and electronic structure of carbon nanotube networks:'super'-carbon nanotubes Journal Article
Em: Nanotechnology, vol. 17, não 3, pp. 617, 2006.
Resumo | Links | BibTeX | Tags: DFT, Mechanical Properties, Molecular Dynamics, Super Carbons
@article{coluci2006geometric,
title = {Geometric and electronic structure of carbon nanotube networks:'super'-carbon nanotubes},
author = {Coluci, Vitor R and Galvao, Douglas S and Jorio, A},
url = {http://iopscience.iop.org/0957-4484/17/3/001},
year = {2006},
date = {2006-01-01},
journal = {Nanotechnology},
volume = {17},
number = {3},
pages = {617},
publisher = {IoP Publishing},
abstract = {Structures of the so-called super-carbon nanotubes are proposed. These structures are built from single walled carbon nanotubes connected by Y-like junctions forming a 'super'-sheet that is then rolled into a seamless cylinder. Such a procedure can be repeated several times, generating a fractal structure. This procedure is not limited to carbon nanotubes, and can be easily modified for application to other systems. Tight binding total energy and density of states calculations showed that the 'super'-sheets and tubes are stable and predicted to present metallic and semiconducting behaviour.},
keywords = {DFT, Mechanical Properties, Molecular Dynamics, Super Carbons},
pubstate = {published},
tppubtype = {article}
}

Rurali, R; Coluci, VR; Galvao, DS
Prediction of Giant Electro-actuation for Carbon Nanoscrolls Journal Article
Em: arXiv preprint cond-mat/0603239, 2006.
Resumo | Links | BibTeX | Tags: DFT, Electroactuation, Electronic Structure, Scrolls
@article{rurali2006predictionb,
title = {Prediction of Giant Electro-actuation for Carbon Nanoscrolls},
author = {Rurali, R and Coluci, VR and Galvao, DS},
url = {http://arxiv.org/abs/cond-mat/0603239},
year = {2006},
date = {2006-01-01},
journal = {arXiv preprint cond-mat/0603239},
abstract = {We study by first-principles calculations the electro-mechanical response of carbon nanoscrolls. We show that although they present a very similar behavior to carbon nanotubes for what concerns the axial deformation sensitivity, they exhibit a radial response upon charge injection which is up to one order of magnitude larger. In association with their high stability, this behavior make them a natural choice for a new class of very efficient nano-actuators.},
keywords = {DFT, Electroactuation, Electronic Structure, Scrolls},
pubstate = {published},
tppubtype = {article}
}
Rurali, R; Coluci, VR; Galvao, DS
Prediction of giant electroactuation for papyruslike carbon nanoscroll structures: first-principles calculations Journal Article
Em: Physical Review B, vol. 74, não 8, pp. 085414, 2006.
Resumo | Links | BibTeX | Tags: DFT, Electronic Structure, Eletroactuation, Scrolls
@article{rurali2006prediction,
title = {Prediction of giant electroactuation for papyruslike carbon nanoscroll structures: first-principles calculations},
author = {Rurali, R and Coluci, VR and Galvao, DS},
url = {http://journals.aps.org/prb/abstract/10.1103/PhysRevB.74.085414},
year = {2006},
date = {2006-01-01},
journal = {Physical Review B},
volume = {74},
number = {8},
pages = {085414},
publisher = {American Physical Society},
abstract = {We study by first-principles calculations the electromechanical response of carbon nanoscroll structures. We show that although they present a very similar behavior to carbon nanotubes in their axial deformation sensitivity, they exhibit a radial response upon charge injection which is up to one order of magnitude larger. In association with their high stability, this behavior makes them a natural choice for a new class of very efficient nanoactuators.},
keywords = {DFT, Electronic Structure, Eletroactuation, Scrolls},
pubstate = {published},
tppubtype = {article}
}
2005
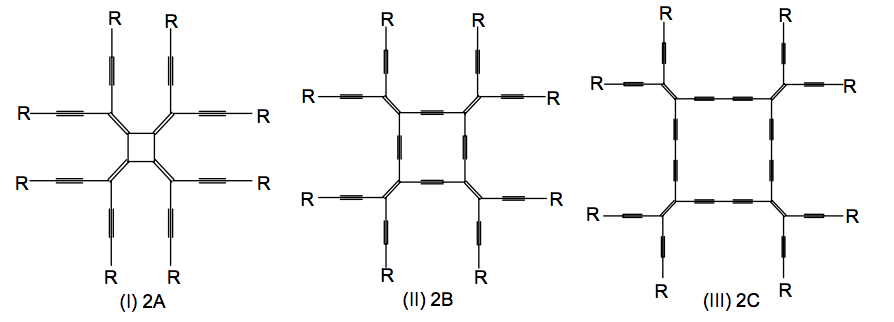
Konstantinova, Elena; Galvao, Douglas S; Barone, Paulo MVB; Dantas, Socrates O
Structural and electronic properties of radialenes and related systems Journal Article
Em: Journal of Molecular Structure: THEOCHEM, vol. 729, não 3, pp. 203–210, 2005.
Resumo | Links | BibTeX | Tags: DFT, Electronic Structure, Radialenes
@article{konstantinova2005structural,
title = {Structural and electronic properties of radialenes and related systems},
author = {Konstantinova, Elena and Galvao, Douglas S and Barone, Paulo MVB and Dantas, Socrates O},
url = {http://www.sciencedirect.com/science/article/pii/S016612800500480X},
year = {2005},
date = {2005-01-01},
journal = {Journal of Molecular Structure: THEOCHEM},
volume = {729},
number = {3},
pages = {203--210},
publisher = {Elsevier},
abstract = {The discovery of new allotropic forms of carbon gives rise to a great interest in carbon compounds as building blocks for novel nanostructure materials. Radialenes are homologous series of compounds with a cycloalkane nucleus bound to methylene side groups, with molecular formula CnHn. The series of expanded radialenes of molecular formulae C2nHn and C3nHn are obtained by inserting acetylene or diacetylene groups between each pair of methylene units. This paper is a report on the theoretical study of structural, electronic and spectroscopic properties of radialenes, expanded radialenes and related molecular systems. Using semiempirical methods we explore the behavior of π-electrons along the carbon-rich skeleton. The results for structural parameters are in a good agreement with the available experimental data. The calculated electronic gaps and spatial distribution of frontier orbitals indicate to interesting electrical and nonlinear optical properties of the explored compounds, which may be useful for technological applications.},
keywords = {DFT, Electronic Structure, Radialenes},
pubstate = {published},
tppubtype = {article}
}
2004
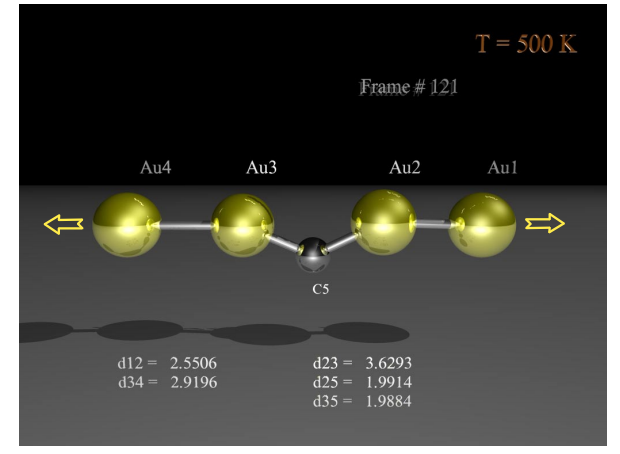
Legoas, Sergio B; Rodrigues, Varlei; Ugarte, Daniel; Galvao, Douglas S
Contaminants in suspended gold chains: An ab initio molecular dynamics study Journal Article
Em: Physical Review Letters, vol. 93, não 21, pp. 216103, 2004.
Resumo | Links | BibTeX | Tags: DFT, Electronic Structure, Linear Atomic Chains, Metallic Nanowires, top20
@article{legoas2004contaminants,
title = {Contaminants in suspended gold chains: An ab initio molecular dynamics study},
author = {Legoas, Sergio B and Rodrigues, Varlei and Ugarte, Daniel and Galvao, Douglas S},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.93.216103},
year = {2004},
date = {2004-01-01},
journal = {Physical Review Letters},
volume = {93},
number = {21},
pages = {216103},
publisher = {American Physical Society},
abstract = {Recently, we have proposed that the origin of anomalously long interatomic distances in suspended gold chains could be the result of carbon contamination during sample manipulation [S. B. Legoas et al., Phys. Rev. Lett. 88, 076105 (2002)]. More recently, however, other works have proposed that hydrogen instead of carbon should be the most probable contaminant. We report ab initio molecular dynamics results for different temperatures considering different possible contaminants. Our results show that at nonzero temperatures (more realistic to simulate the experimental conditions) hydrogen may be ruled out and carbon atoms remain the best candidate for contamination.},
keywords = {DFT, Electronic Structure, Linear Atomic Chains, Metallic Nanowires, top20},
pubstate = {published},
tppubtype = {article}
}
2002

Legoas, Sergio B; Galvao, Douglas S; Rodrigues, Varlei; Ugarte, Daniel
Origin of anomalously long interatomic distances in suspended gold chains Journal Article
Em: Physical Review Letters, vol. 88, não 7, pp. 076105, 2002.
Resumo | Links | BibTeX | Tags: DFT, Electronic Structure, Linear Atomic Chains, Metallic Nanowires, TEM, top20
@article{legoas2002origin,
title = {Origin of anomalously long interatomic distances in suspended gold chains},
author = {Legoas, Sergio B and Galvao, Douglas S and Rodrigues, Varlei and Ugarte, Daniel},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.88.076105},
year = {2002},
date = {2002-01-01},
journal = {Physical Review Letters},
volume = {88},
number = {7},
pages = {076105},
publisher = {American Physical Society},
abstract = {The discovery of long bonds in gold atom chains has represented a challenge for physical interpretation. In fact, interatomic distances frequently attain 3.0–3.6 Å values, and distances as large as 5.0 Å may be occasionally observed. Here we studied gold chains by transmission electron microscopy and performed theoretical calculations using cluster ab initio density functional formalism. We show that the insertion of two carbon atoms is required to account for the longest bonds, while distances above 3 Å may be due to a mixture of clean and one C atom contaminated bonds.},
keywords = {DFT, Electronic Structure, Linear Atomic Chains, Metallic Nanowires, TEM, top20},
pubstate = {published},
tppubtype = {article}
}
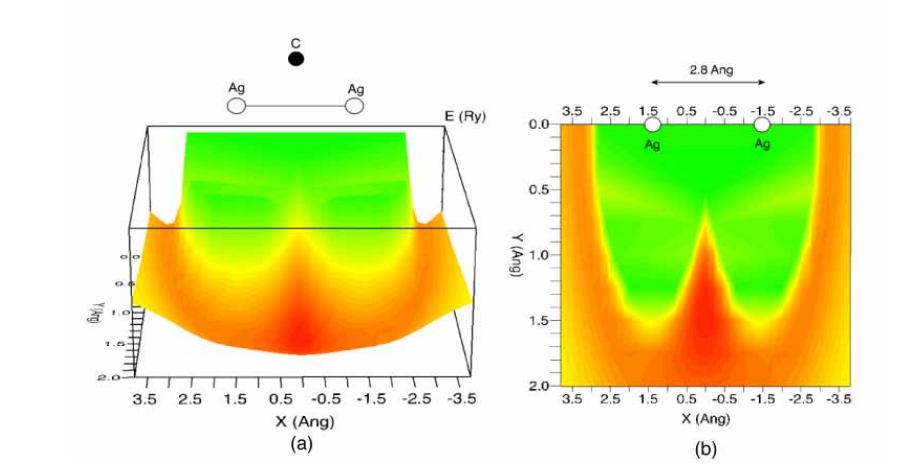
Legoas, Sergio B; Galvao, Douglas S; Rodrigues, Varlei; Ugarte, Daniel
The Role of Carbon Contamination in Suspended Gold Nanowires Journal Article
Em: MRS Proceedings, vol. 738, pp. G14–6, 2002.
Resumo | Links | BibTeX | Tags: DFT, Electronic Structure, Linear Atomic Chains, Metallic Nanowires, TEM
@article{legoas2002role,
title = {The Role of Carbon Contamination in Suspended Gold Nanowires},
author = {Legoas, Sergio B and Galvao, Douglas S and Rodrigues, Varlei and Ugarte, Daniel},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8031754&fileId=S1946427400148249},
year = {2002},
date = {2002-01-01},
journal = {MRS Proceedings},
volume = {738},
pages = {G14--6},
publisher = {Cambridge University Press},
abstract = {Metallic nanowires represent very interesting systems due to new phenomena such as quantum conductance and unexpected long interatomic distances attaining 0.3–0.5 nm. These large distances represent a challenge for physical interpretation. In this work we present experimental data from transmission electron microscopy and results from ab initio density functional calculations for suspended gold chains. We show that large distances as 0.5 nm can be easily explained by the presence of carbon atoms as contaminants, while distances ranging from 0.29 up to 0.36 nm might be explained as resulting of a mixture of clean stressed and contaminated linear chains.},
keywords = {DFT, Electronic Structure, Linear Atomic Chains, Metallic Nanowires, TEM},
pubstate = {published},
tppubtype = {article}
}

