
Ok-Kyung; Owuor Park, Peter; Morais Jaques
Novel Method to Fabricate Multi-Functional Boron Nitride-Iron-Carbon Nanotube Hybrid Materials for Fabrication of High- Performance Polyimide Composites (under review) Journal Article
In: 2019.
@article{Park2019,
title = {Novel Method to Fabricate Multi-Functional Boron Nitride-Iron-Carbon Nanotube Hybrid Materials for Fabrication of High- Performance Polyimide Composites (under review)},
author = {Park, Ok-Kyung; Owuor, Peter; Morais Jaques, Ygor; Lee, Joong Hee; Kim, Nam
Hoon; Galvao, Douglas; Lou, Jun; Tiwary, Chandra; Ajayan, Pulickel},
year = {2019},
date = {2019-01-05},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Perim, Eric; Paupitz, Ricardo; Galvao, Douglas S
Controlled route to the fabrication of carbon and boron nitride nanoscrolls: A molecular dynamics investigation Journal Article
In: Journal of Applied Physics, vol. 113, no. 5, pp. 054306, 2013.
@article{perim2013controlled,
title = {Controlled route to the fabrication of carbon and boron nitride nanoscrolls: A molecular dynamics investigation},
author = {Perim, Eric and Paupitz, Ricardo and Galvao, Douglas S},
url = {http://scitation.aip.org/content/aip/journal/jap/113/5/10.1063/1.4790304},
year = {2013},
date = {2013-01-01},
journal = {Journal of Applied Physics},
volume = {113},
number = {5},
pages = {054306},
publisher = {AIP Publishing},
abstract = {Carbon nanoscrolls (graphene layers rolled up into papyrus-like tubular structures) are nanostructures with unique and interesting characteristics that could be exploited to build several new nanodevices. However, an efficient and controlled synthesis of these structures was not achieved yet, making its large scale production a challenge to materials scientists. Also, the formation process and detailed mechanisms that occur during its synthesis are not completely known. In this work, using fully atomistic molecular dynamics simulations, we discuss a possible route to nanoscrolls made from graphene layers deposited over silicon oxide substrates containing chambers/pits. The scrolling mechanism is triggered by carbon nanotubes deposited on the layers. The process is completely general and can be used to produce scrolls from other lamellar materials, like boron nitride, for instance.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Perim, E; Autreto, PAS; Paupitz, R; Galvao, DS
Dynamical aspects of the unzipping of multiwalled boron nitride nanotubes Journal Article
In: Physical Chemistry Chemical Physics, vol. 15, no. 44, pp. 19147–19150, 2013.
@article{perim2013dynamical,
title = {Dynamical aspects of the unzipping of multiwalled boron nitride nanotubes},
author = {Perim, E and Autreto, PAS and Paupitz, R and Galvao, DS},
url = {http://pubs.rsc.org/EN/content/articlehtml/2013/cp/c3cp52701h},
year = {2013},
date = {2013-01-01},
journal = {Physical Chemistry Chemical Physics},
volume = {15},
number = {44},
pages = {19147--19150},
publisher = {Royal Society of Chemistry},
abstract = {Boron nitride nanoribbons (BNNRs) exhibit very interesting magnetic properties, which could be very useful in the development of spintronic based devices. One possible route to obtain BNNRs is through the unzipping of boron nitride nanotubes (BNNTs), which have been already experimentally realized. In this work, different aspects of the unzipping process of BNNTs were investigated through fully atomistic molecular dynamics simulations using a classical reactive force field (ReaxFF). We investigated multiwalled BNNTs of different diameters and chiralities. Our results show that chirality plays a very important role in the unzipping process, as well as the interlayer coupling. These combined aspects significantly change the fracturing patterns and several other features of the unzipping processes in comparison to the ones observed for carbon nanotubes. Also, similar to carbon nanotubes, defective BNNTs can create regions of very high curvature which can act as a path to the unzipping process.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Perim, Eric; Santos, Ricardo Paupitz; Autreto, Pedro Alves da Silva; Galvao, Douglas S
Fracture Patterns of Boron Nitride Nanotubes Proceedings
Cambridge University Press, vol. 1526, 2013.
@proceedings{perim2013fracture,
title = {Fracture Patterns of Boron Nitride Nanotubes},
author = {Perim, Eric and Santos, Ricardo Paupitz and Autreto, Pedro Alves da Silva and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8883390&fileId=S1946427413004946},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1526},
pages = {mrsf12--1526},
publisher = {Cambridge University Press},
abstract = {During the last years carbon-based nanostructures (such as, fullerenes, carbon nanotubes and graphene) have been object of intense investigations. The great interest in these nanostructures can be attributed to their remarkable electrical and mechanical properties. Their inorganic equivalent structures do exist and are based on boron nitride (BN) motifs. BN fullerenes, nanotubes and single layers have been already synthesized. Recently, the fracture patterns of single layer graphene and multi-walled carbon nanotubes under stress have been studied by theoretical and experimental methods. In this work we investigated the fracturing process of defective carbon and boron nitride nanotubes under similar stress conditions. We have carried out fully atomistic molecular reactive molecular dynamics simulations using the ReaxFF force field. The similarities and differences between carbon and boron nitride fracture patterns are addressed.},
keywords = {},
pubstate = {published},
tppubtype = {proceedings}
}
Perim, Eric; Paupitz, Ricardo; Autreto, PAS; Galvao, DS
The Hydrogenation Dynamics of h-BN Sheets Proceedings
Cambridge University Press, vol. 1549, 2013.
@proceedings{perim2013hydrogenation,
title = {The Hydrogenation Dynamics of h-BN Sheets},
author = {Perim, Eric and Paupitz, Ricardo and Autreto, PAS and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8943477&fileId=S1946427413007938},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1549},
pages = {91--98},
publisher = {Cambridge University Press},
abstract = {Hexagonal boron nitride (h-BN), also known as white graphite, is the inorganic analogue of graphite. Single layers of both structures have been already experimentally realized.
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics of hydrogenation of h-BN single-layers membranes.
Our results show that the rate of hydrogenation atoms bonded to the membrane is highly dependent on the temperature and that only at low temperatures there is a preferential bond to boron atoms. Unlike graphanes (hydrogenated graphene), hydrogenated h-BN membranes do not exhibit the formation of correlated domains. Also, the out-of-plane deformations are more pronounced in comparison with the graphene case. After a critical number of incorporated hydrogen atoms the membrane become increasingly defective, lost its two-dimensional character and collapses. The hydrogen radial pair distribution and second-nearest neighbor correlations were also analyzed.},
keywords = {},
pubstate = {published},
tppubtype = {proceedings}
}
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics of hydrogenation of h-BN single-layers membranes.
Our results show that the rate of hydrogenation atoms bonded to the membrane is highly dependent on the temperature and that only at low temperatures there is a preferential bond to boron atoms. Unlike graphanes (hydrogenated graphene), hydrogenated h-BN membranes do not exhibit the formation of correlated domains. Also, the out-of-plane deformations are more pronounced in comparison with the graphene case. After a critical number of incorporated hydrogen atoms the membrane become increasingly defective, lost its two-dimensional character and collapses. The hydrogen radial pair distribution and second-nearest neighbor correlations were also analyzed.
Perim, E; Galvao, DS
Boron Nitride Nanoscrolls Journal Article
In: Physicæ Proceedings, vol. 1, no. 1, pp. 2, 2012.
@article{perim2012boron,
title = {Boron Nitride Nanoscrolls},
author = {Perim, E and Galvao, DS},
url = {http://physicae.ifi.unicamp.br/phyproceedings/article/view/269},
year = {2012},
date = {2012-01-01},
journal = {Physicæ Proceedings},
volume = {1},
number = {1},
pages = {2},
abstract = {Recently, based on computer simulations, it has been proposed that stable boron nitride nanoscrolls (BNNSs) can exist. In this work we show that the BNNSs stability mechanisms follow the same simple physical principles proposed for carbon nanoscrolls (CNSs). For both classes of scrolls, the mechanical stability arises as the result of the interplay between attractive van der Waals forces and the elastic (bending) deformations. The topology (chirality) of the scrolled single-layer membranes plays an important role defining BNNS stability. A controled way to produce BNNSs is also addressed.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
dos Santos, Ricardo P; Machado, Leonardo D; Legoas, Sergio B; Galvao, Douglas S
Tribological Properties of Graphene and Boron-Nitride Layers: A Fully Atomistic Molecular Dynamics Study Proceedings
Cambridge University Press, vol. 1407, 2012.
@proceedings{dos2012tribological,
title = {Tribological Properties of Graphene and Boron-Nitride Layers: A Fully Atomistic Molecular Dynamics Study},
author = {dos Santos, Ricardo P and Machado, Leonardo D and Legoas, Sergio B and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8537106&fileId=S1946427412007063},
year = {2012},
date = {2012-01-01},
journal = {MRS Proceedings},
volume = {1407},
pages = {mrsf11--1407},
publisher = {Cambridge University Press},
abstract = {Graphene has been one of the most important subjects in materials science in the last years. Recently, the frictional characteristics of atomically thin sheets were experimentally investigated using atomic force microscopy (AFM). A new mechanism to explain the enhanced friction for these materials, based on elastic compliance has been proposed. Here, we have investigated the tribological properties of graphene and boron-nitride (single and multi-layers) membranes using fully atomistic molecular dynamics simulations. These simulations were carried out using classical force fields, as implemented in the Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) code. The used structural models contain typically hundreds of thousands of atoms. In order to mimic the experimental conditions, an artificial AFM tip was moved over the membranes and the tribological characteristics determined in terms of forces and energies. Our results are in good agreement with the available experimental data. They show that the observed enhanced tribological properties can be explained in terms of out-of-plane geometrical distortions and elastic waves propagation. They validate the general features of the model proposed by Lee et al. (Science 328, 76 (2010).},
keywords = {},
pubstate = {published},
tppubtype = {proceedings}
}
Perim, Eric; Galvao, Douglas S
Stability and Dynamics of Boron Nitride Nanoscrolls Proceedings
Cambridge University Press, vol. 1307, 2011.
@proceedings{perim2011stability,
title = {Stability and Dynamics of Boron Nitride Nanoscrolls},
author = {Perim, Eric and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayFulltext?type=1&fid=8200678&jid=OPL&volumeId=1307&issueId=-1&aid=8200676},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1307},
pages = {mrsf10--1307},
publisher = {Cambridge University Press},
abstract = {We report here molecular dynamics results for boron nitride nanoscroll structures
(BNNSs) with relation to their stability and formation mechanisms. We show that, similarly to
carbon nanoscrolls, BNNSs are stable due to van der Waals interactions among overlapping
layers. The energy balance between losses and gains (due to elastic deformations and van der
Waals interactions, respectively) when the structure is rolled up leads to the existence of a
critical value of the internal scroll diameter where stable or metastable structures can be formed.
The mechanisms of scroll formation and stability as a function of their chirality were also
investigated.},
keywords = {},
pubstate = {published},
tppubtype = {proceedings}
}
(BNNSs) with relation to their stability and formation mechanisms. We show that, similarly to
carbon nanoscrolls, BNNSs are stable due to van der Waals interactions among overlapping
layers. The energy balance between losses and gains (due to elastic deformations and van der
Waals interactions, respectively) when the structure is rolled up leads to the existence of a
critical value of the internal scroll diameter where stable or metastable structures can be formed.
The mechanisms of scroll formation and stability as a function of their chirality were also
investigated.
Garcez, Karl M; Moreira, Edvan; Azevedo, David L; Galvao, Douglas S
Neon atoms oscillating inside carbon and boron nitride nanotubes: a fully atomistic molecular dynamics investigation Journal Article
In: Molecular Simulation, vol. 36, no. 9, pp. 639–643, 2010.
@article{garcez2010neon,
title = {Neon atoms oscillating inside carbon and boron nitride nanotubes: a fully atomistic molecular dynamics investigation},
author = {Garcez, Karl M and Moreira, Edvan and Azevedo, David L and Galvao, Douglas S},
url = {http://www.tandfonline.com/doi/abs/10.1080/08927020903463926#.VLfp54rF-2o},
year = {2010},
date = {2010-01-01},
journal = {Molecular Simulation},
volume = {36},
number = {9},
pages = {639--643},
publisher = {Taylor & Francis Group},
abstract = {In the present work, based on extensive fully atomistic molecular dynamics simulations, we discuss the dynamics of neon atoms oscillating inside (5,5) single-walled carbon nanotubes (CNTs) and boron nitride nanotubes (BNNTs). Our results show that sustained high-frequency oscillatory regimes are possible for a large range of temperatures. Our results also show that the general features of the oscillations are quite similar to those observed in CNT and BNNT, in contrast with some speculations in previous works in the literature about the importance of broken symmetry and chirality exhibited by BNNTs.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Perim, Eric; Galvao, Douglas S
The structure and dynamics of boron nitride nanoscrolls Journal Article
In: Nanotechnology, vol. 20, no. 33, pp. 335702, 2009.
@article{perim2009structure,
title = {The structure and dynamics of boron nitride nanoscrolls},
author = {Perim, Eric and Galvao, Douglas S},
url = {http://iopscience.iop.org/0957-4484/20/33/335702},
year = {2009},
date = {2009-01-01},
journal = {Nanotechnology},
volume = {20},
number = {33},
pages = {335702},
publisher = {IOP Publishing},
abstract = {Carbon nanoscrolls (CNSs) are structures formed by rolling up graphene layers into a scroll-like shape. CNNs have been experimentally produced by different groups. Boron nitride nanoscrolls (BNNSs) are similar structures using boron nitride instead of graphene layers. In this paper we report molecular mechanics and molecular dynamics results for the structural and dynamical aspects of BNNS formation. Similarly to CNS, BNNS formation is dominated by two major energy contributions, the increase in the elastic energy and the energetic gain due to van der Waals interactions of the overlapping surface of the rolled layers. The armchair scrolls are the most stable configuration while zigzag scrolls are metastable structures which can be thermally converted to armchairs. Chiral scrolls are unstable and tend to evolve into zigzag or armchair configurations depending on their initial geometries. The possible experimental routes to produce BNNSs are also addressed.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2019
Ok-Kyung; Owuor Park, Peter; Morais Jaques
Novel Method to Fabricate Multi-Functional Boron Nitride-Iron-Carbon Nanotube Hybrid Materials for Fabrication of High- Performance Polyimide Composites (under review) Journal Article
In: 2019.
BibTeX | Tags: Boron Nitride, carbon nanotube, Modeling
@article{Park2019,
title = {Novel Method to Fabricate Multi-Functional Boron Nitride-Iron-Carbon Nanotube Hybrid Materials for Fabrication of High- Performance Polyimide Composites (under review)},
author = {Park, Ok-Kyung; Owuor, Peter; Morais Jaques, Ygor; Lee, Joong Hee; Kim, Nam
Hoon; Galvao, Douglas; Lou, Jun; Tiwary, Chandra; Ajayan, Pulickel},
year = {2019},
date = {2019-01-05},
keywords = {Boron Nitride, carbon nanotube, Modeling},
pubstate = {published},
tppubtype = {article}
}
2013

Perim, Eric; Paupitz, Ricardo; Galvao, Douglas S
Controlled route to the fabrication of carbon and boron nitride nanoscrolls: A molecular dynamics investigation Journal Article
In: Journal of Applied Physics, vol. 113, no. 5, pp. 054306, 2013.
Abstract | Links | BibTeX | Tags: Boron Nitride, Carbon Nanotubes, Graphene, Molecular Dynamics, Scrolls
@article{perim2013controlled,
title = {Controlled route to the fabrication of carbon and boron nitride nanoscrolls: A molecular dynamics investigation},
author = {Perim, Eric and Paupitz, Ricardo and Galvao, Douglas S},
url = {http://scitation.aip.org/content/aip/journal/jap/113/5/10.1063/1.4790304},
year = {2013},
date = {2013-01-01},
journal = {Journal of Applied Physics},
volume = {113},
number = {5},
pages = {054306},
publisher = {AIP Publishing},
abstract = {Carbon nanoscrolls (graphene layers rolled up into papyrus-like tubular structures) are nanostructures with unique and interesting characteristics that could be exploited to build several new nanodevices. However, an efficient and controlled synthesis of these structures was not achieved yet, making its large scale production a challenge to materials scientists. Also, the formation process and detailed mechanisms that occur during its synthesis are not completely known. In this work, using fully atomistic molecular dynamics simulations, we discuss a possible route to nanoscrolls made from graphene layers deposited over silicon oxide substrates containing chambers/pits. The scrolling mechanism is triggered by carbon nanotubes deposited on the layers. The process is completely general and can be used to produce scrolls from other lamellar materials, like boron nitride, for instance.},
keywords = {Boron Nitride, Carbon Nanotubes, Graphene, Molecular Dynamics, Scrolls},
pubstate = {published},
tppubtype = {article}
}

Perim, E; Autreto, PAS; Paupitz, R; Galvao, DS
Dynamical aspects of the unzipping of multiwalled boron nitride nanotubes Journal Article
In: Physical Chemistry Chemical Physics, vol. 15, no. 44, pp. 19147–19150, 2013.
Abstract | Links | BibTeX | Tags: Boron Nitride, Mechanical Properties, Molecular Dynamics, Unzipping
@article{perim2013dynamical,
title = {Dynamical aspects of the unzipping of multiwalled boron nitride nanotubes},
author = {Perim, E and Autreto, PAS and Paupitz, R and Galvao, DS},
url = {http://pubs.rsc.org/EN/content/articlehtml/2013/cp/c3cp52701h},
year = {2013},
date = {2013-01-01},
journal = {Physical Chemistry Chemical Physics},
volume = {15},
number = {44},
pages = {19147--19150},
publisher = {Royal Society of Chemistry},
abstract = {Boron nitride nanoribbons (BNNRs) exhibit very interesting magnetic properties, which could be very useful in the development of spintronic based devices. One possible route to obtain BNNRs is through the unzipping of boron nitride nanotubes (BNNTs), which have been already experimentally realized. In this work, different aspects of the unzipping process of BNNTs were investigated through fully atomistic molecular dynamics simulations using a classical reactive force field (ReaxFF). We investigated multiwalled BNNTs of different diameters and chiralities. Our results show that chirality plays a very important role in the unzipping process, as well as the interlayer coupling. These combined aspects significantly change the fracturing patterns and several other features of the unzipping processes in comparison to the ones observed for carbon nanotubes. Also, similar to carbon nanotubes, defective BNNTs can create regions of very high curvature which can act as a path to the unzipping process.
},
keywords = {Boron Nitride, Mechanical Properties, Molecular Dynamics, Unzipping},
pubstate = {published},
tppubtype = {article}
}

Perim, Eric; Santos, Ricardo Paupitz; Autreto, Pedro Alves da Silva; Galvao, Douglas S
Fracture Patterns of Boron Nitride Nanotubes Proceedings
Cambridge University Press, vol. 1526, 2013.
Abstract | Links | BibTeX | Tags: Boron Nitride, Fracture, Mechanical Properties, Unzipping
@proceedings{perim2013fracture,
title = {Fracture Patterns of Boron Nitride Nanotubes},
author = {Perim, Eric and Santos, Ricardo Paupitz and Autreto, Pedro Alves da Silva and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8883390&fileId=S1946427413004946},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1526},
pages = {mrsf12--1526},
publisher = {Cambridge University Press},
abstract = {During the last years carbon-based nanostructures (such as, fullerenes, carbon nanotubes and graphene) have been object of intense investigations. The great interest in these nanostructures can be attributed to their remarkable electrical and mechanical properties. Their inorganic equivalent structures do exist and are based on boron nitride (BN) motifs. BN fullerenes, nanotubes and single layers have been already synthesized. Recently, the fracture patterns of single layer graphene and multi-walled carbon nanotubes under stress have been studied by theoretical and experimental methods. In this work we investigated the fracturing process of defective carbon and boron nitride nanotubes under similar stress conditions. We have carried out fully atomistic molecular reactive molecular dynamics simulations using the ReaxFF force field. The similarities and differences between carbon and boron nitride fracture patterns are addressed.},
keywords = {Boron Nitride, Fracture, Mechanical Properties, Unzipping},
pubstate = {published},
tppubtype = {proceedings}
}
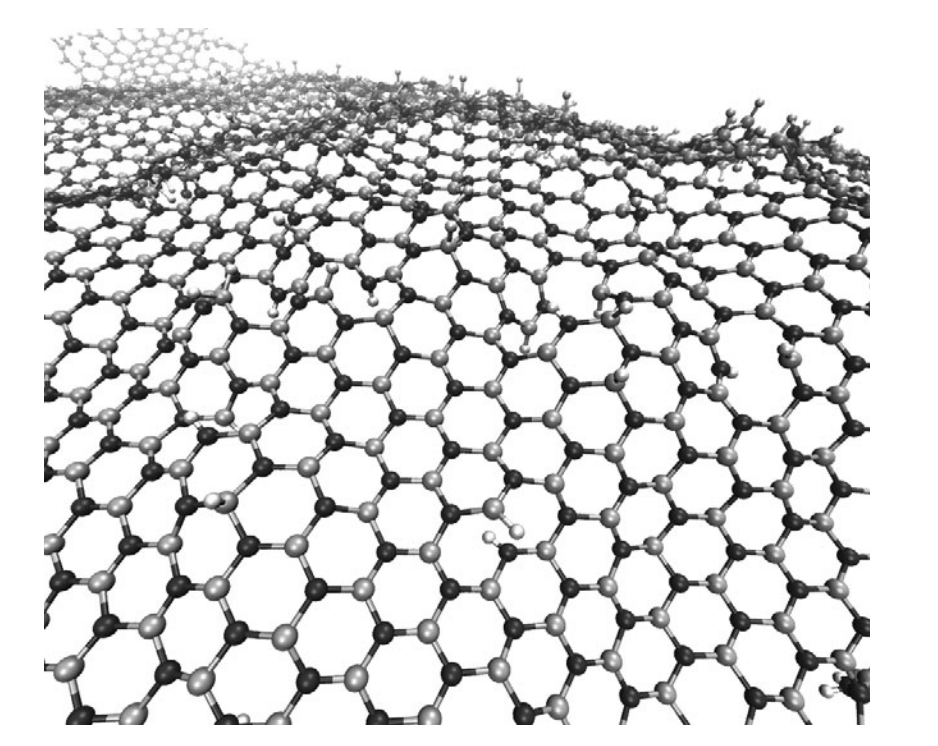
Perim, Eric; Paupitz, Ricardo; Autreto, PAS; Galvao, DS
The Hydrogenation Dynamics of h-BN Sheets Proceedings
Cambridge University Press, vol. 1549, 2013.
Abstract | Links | BibTeX | Tags: Boron Nitride, Hydrogenation, Molecular Dynamics, Nanotubes
@proceedings{perim2013hydrogenation,
title = {The Hydrogenation Dynamics of h-BN Sheets},
author = {Perim, Eric and Paupitz, Ricardo and Autreto, PAS and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8943477&fileId=S1946427413007938},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1549},
pages = {91--98},
publisher = {Cambridge University Press},
abstract = {Hexagonal boron nitride (h-BN), also known as white graphite, is the inorganic analogue of graphite. Single layers of both structures have been already experimentally realized.
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics of hydrogenation of h-BN single-layers membranes.
Our results show that the rate of hydrogenation atoms bonded to the membrane is highly dependent on the temperature and that only at low temperatures there is a preferential bond to boron atoms. Unlike graphanes (hydrogenated graphene), hydrogenated h-BN membranes do not exhibit the formation of correlated domains. Also, the out-of-plane deformations are more pronounced in comparison with the graphene case. After a critical number of incorporated hydrogen atoms the membrane become increasingly defective, lost its two-dimensional character and collapses. The hydrogen radial pair distribution and second-nearest neighbor correlations were also analyzed.},
keywords = {Boron Nitride, Hydrogenation, Molecular Dynamics, Nanotubes},
pubstate = {published},
tppubtype = {proceedings}
}
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics of hydrogenation of h-BN single-layers membranes.
Our results show that the rate of hydrogenation atoms bonded to the membrane is highly dependent on the temperature and that only at low temperatures there is a preferential bond to boron atoms. Unlike graphanes (hydrogenated graphene), hydrogenated h-BN membranes do not exhibit the formation of correlated domains. Also, the out-of-plane deformations are more pronounced in comparison with the graphene case. After a critical number of incorporated hydrogen atoms the membrane become increasingly defective, lost its two-dimensional character and collapses. The hydrogen radial pair distribution and second-nearest neighbor correlations were also analyzed.
2012
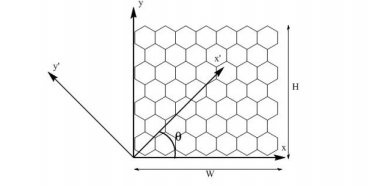
Perim, E; Galvao, DS
Boron Nitride Nanoscrolls Journal Article
In: Physicæ Proceedings, vol. 1, no. 1, pp. 2, 2012.
Abstract | Links | BibTeX | Tags: Boron Nitride, Molecular Dynamics, Scrolls
@article{perim2012boron,
title = {Boron Nitride Nanoscrolls},
author = {Perim, E and Galvao, DS},
url = {http://physicae.ifi.unicamp.br/phyproceedings/article/view/269},
year = {2012},
date = {2012-01-01},
journal = {Physicæ Proceedings},
volume = {1},
number = {1},
pages = {2},
abstract = {Recently, based on computer simulations, it has been proposed that stable boron nitride nanoscrolls (BNNSs) can exist. In this work we show that the BNNSs stability mechanisms follow the same simple physical principles proposed for carbon nanoscrolls (CNSs). For both classes of scrolls, the mechanical stability arises as the result of the interplay between attractive van der Waals forces and the elastic (bending) deformations. The topology (chirality) of the scrolled single-layer membranes plays an important role defining BNNS stability. A controled way to produce BNNSs is also addressed.},
keywords = {Boron Nitride, Molecular Dynamics, Scrolls},
pubstate = {published},
tppubtype = {article}
}

dos Santos, Ricardo P; Machado, Leonardo D; Legoas, Sergio B; Galvao, Douglas S
Tribological Properties of Graphene and Boron-Nitride Layers: A Fully Atomistic Molecular Dynamics Study Proceedings
Cambridge University Press, vol. 1407, 2012.
Abstract | Links | BibTeX | Tags: Boron Nitride, Graphene, Molecular Dynamics, Tribology
@proceedings{dos2012tribological,
title = {Tribological Properties of Graphene and Boron-Nitride Layers: A Fully Atomistic Molecular Dynamics Study},
author = {dos Santos, Ricardo P and Machado, Leonardo D and Legoas, Sergio B and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8537106&fileId=S1946427412007063},
year = {2012},
date = {2012-01-01},
journal = {MRS Proceedings},
volume = {1407},
pages = {mrsf11--1407},
publisher = {Cambridge University Press},
abstract = {Graphene has been one of the most important subjects in materials science in the last years. Recently, the frictional characteristics of atomically thin sheets were experimentally investigated using atomic force microscopy (AFM). A new mechanism to explain the enhanced friction for these materials, based on elastic compliance has been proposed. Here, we have investigated the tribological properties of graphene and boron-nitride (single and multi-layers) membranes using fully atomistic molecular dynamics simulations. These simulations were carried out using classical force fields, as implemented in the Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) code. The used structural models contain typically hundreds of thousands of atoms. In order to mimic the experimental conditions, an artificial AFM tip was moved over the membranes and the tribological characteristics determined in terms of forces and energies. Our results are in good agreement with the available experimental data. They show that the observed enhanced tribological properties can be explained in terms of out-of-plane geometrical distortions and elastic waves propagation. They validate the general features of the model proposed by Lee et al. (Science 328, 76 (2010).},
keywords = {Boron Nitride, Graphene, Molecular Dynamics, Tribology},
pubstate = {published},
tppubtype = {proceedings}
}
2011
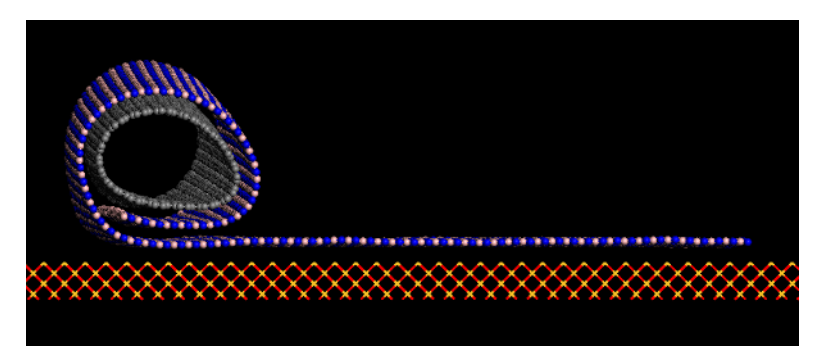
Perim, Eric; Galvao, Douglas S
Stability and Dynamics of Boron Nitride Nanoscrolls Proceedings
Cambridge University Press, vol. 1307, 2011.
Abstract | Links | BibTeX | Tags: Boron Nitride, Molecular Dynamics, Scrolls
@proceedings{perim2011stability,
title = {Stability and Dynamics of Boron Nitride Nanoscrolls},
author = {Perim, Eric and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayFulltext?type=1&fid=8200678&jid=OPL&volumeId=1307&issueId=-1&aid=8200676},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1307},
pages = {mrsf10--1307},
publisher = {Cambridge University Press},
abstract = {We report here molecular dynamics results for boron nitride nanoscroll structures
(BNNSs) with relation to their stability and formation mechanisms. We show that, similarly to
carbon nanoscrolls, BNNSs are stable due to van der Waals interactions among overlapping
layers. The energy balance between losses and gains (due to elastic deformations and van der
Waals interactions, respectively) when the structure is rolled up leads to the existence of a
critical value of the internal scroll diameter where stable or metastable structures can be formed.
The mechanisms of scroll formation and stability as a function of their chirality were also
investigated.},
keywords = {Boron Nitride, Molecular Dynamics, Scrolls},
pubstate = {published},
tppubtype = {proceedings}
}
(BNNSs) with relation to their stability and formation mechanisms. We show that, similarly to
carbon nanoscrolls, BNNSs are stable due to van der Waals interactions among overlapping
layers. The energy balance between losses and gains (due to elastic deformations and van der
Waals interactions, respectively) when the structure is rolled up leads to the existence of a
critical value of the internal scroll diameter where stable or metastable structures can be formed.
The mechanisms of scroll formation and stability as a function of their chirality were also
investigated.
2010
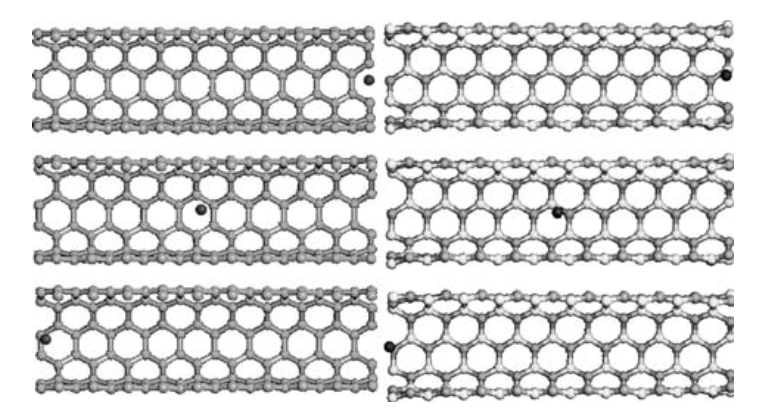
Garcez, Karl M; Moreira, Edvan; Azevedo, David L; Galvao, Douglas S
Neon atoms oscillating inside carbon and boron nitride nanotubes: a fully atomistic molecular dynamics investigation Journal Article
In: Molecular Simulation, vol. 36, no. 9, pp. 639–643, 2010.
Abstract | Links | BibTeX | Tags: Boron Nitride, Encapsulation, Molecular Dynamics, Nanotubes
@article{garcez2010neon,
title = {Neon atoms oscillating inside carbon and boron nitride nanotubes: a fully atomistic molecular dynamics investigation},
author = {Garcez, Karl M and Moreira, Edvan and Azevedo, David L and Galvao, Douglas S},
url = {http://www.tandfonline.com/doi/abs/10.1080/08927020903463926#.VLfp54rF-2o},
year = {2010},
date = {2010-01-01},
journal = {Molecular Simulation},
volume = {36},
number = {9},
pages = {639--643},
publisher = {Taylor & Francis Group},
abstract = {In the present work, based on extensive fully atomistic molecular dynamics simulations, we discuss the dynamics of neon atoms oscillating inside (5,5) single-walled carbon nanotubes (CNTs) and boron nitride nanotubes (BNNTs). Our results show that sustained high-frequency oscillatory regimes are possible for a large range of temperatures. Our results also show that the general features of the oscillations are quite similar to those observed in CNT and BNNT, in contrast with some speculations in previous works in the literature about the importance of broken symmetry and chirality exhibited by BNNTs.},
keywords = {Boron Nitride, Encapsulation, Molecular Dynamics, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
2009

Perim, Eric; Galvao, Douglas S
The structure and dynamics of boron nitride nanoscrolls Journal Article
In: Nanotechnology, vol. 20, no. 33, pp. 335702, 2009.
Abstract | Links | BibTeX | Tags: Boron Nitride, Molecular Dynamics, Scrolls
@article{perim2009structure,
title = {The structure and dynamics of boron nitride nanoscrolls},
author = {Perim, Eric and Galvao, Douglas S},
url = {http://iopscience.iop.org/0957-4484/20/33/335702},
year = {2009},
date = {2009-01-01},
journal = {Nanotechnology},
volume = {20},
number = {33},
pages = {335702},
publisher = {IOP Publishing},
abstract = {Carbon nanoscrolls (CNSs) are structures formed by rolling up graphene layers into a scroll-like shape. CNNs have been experimentally produced by different groups. Boron nitride nanoscrolls (BNNSs) are similar structures using boron nitride instead of graphene layers. In this paper we report molecular mechanics and molecular dynamics results for the structural and dynamical aspects of BNNS formation. Similarly to CNS, BNNS formation is dominated by two major energy contributions, the increase in the elastic energy and the energetic gain due to van der Waals interactions of the overlapping surface of the rolled layers. The armchair scrolls are the most stable configuration while zigzag scrolls are metastable structures which can be thermally converted to armchairs. Chiral scrolls are unstable and tend to evolve into zigzag or armchair configurations depending on their initial geometries. The possible experimental routes to produce BNNSs are also addressed.
},
keywords = {Boron Nitride, Molecular Dynamics, Scrolls},
pubstate = {published},
tppubtype = {article}
}
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ

