
Anna Kremen Nitzan Shadmi, Yiftach Frenkel; Joselevich, Ernesto
Defect-Free Carbon Nanotube Coils Journal Article
In: Nano Letters, vol. 16, no. 4, pp. 2152–2158, 2016.
@article{Shadmi2016,
title = {Defect-Free Carbon Nanotube Coils},
author = {Nitzan Shadmi, Anna Kremen, Yiftach Frenkel, Zachary J. Lapin, Leonardo D. Machado, Sergio B. Legoas, Ora Bitton, Katya Rechav, Ronit Popovitz-Biro, Douglas S. Galvão, Ado Jorio, Lukas Novotny, Beena Kalisky, and Ernesto Joselevich},
url = {http://pubs.acs.org/doi/abs/10.1021/acs.nanolett.5b03417},
doi = {10.1021/acs.nanolett.5b03417},
year = {2016},
date = {2016-04-01},
journal = {Nano Letters},
volume = {16},
number = {4},
pages = {2152–2158},
abstract = {Carbon nanotubes are promising building blocks for various nanoelectronic components. A highly desirable geometry for such applications is a coil. However, coiled nanotube structures reported so far were inherently defective or had no free ends accessible for contacting. Here we demonstrate the spontaneous self-coiling of single-wall carbon nanotubes into defect-free coils of up to more than 70 turns with identical diameter and chirality, and free ends. We characterize the structure, formation mechanism, and electrical properties of these coils by different microscopies, molecular dynamics simulations, Raman spectroscopy, and electrical and magnetic measurements. The coils are highly conductive, as expected for defect-free carbon nanotubes, but adjacent nanotube segments in the coil are more highly coupled than in regular bundles of single-wall carbon nanotubes, owing to their perfect crystal momentum matching, which enables tunneling between the turns. Although this behavior does not yet enable the performance of these nanotube coils as inductive devices, it does point a clear path for their realization. Hence, this study represents a major step toward the production of many different nanotube coil devices, including inductors, electromagnets, transformers, and dynamos.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Gustavo Brunetto Sehmus Ozden, N. S. Karthiselva
Controlled 3D Carbon Nanotube Structures by Plasma Welding Journal Article
In: Advanced Materials Interfaces, vol. 2016, pp. 1500755, 2016.
@article{Ozden2016,
title = {Controlled 3D Carbon Nanotube Structures by Plasma Welding},
author = {Sehmus Ozden, Gustavo Brunetto, N. S. Karthiselva, Douglas S. Galvão, Ajit Roy, Srinivasa R. Bakshi, Chandra S. Tiwary, andPulickel M. Ajayan},
url = {http://onlinelibrary.wiley.com/doi/10.1002/admi.201500755/abstract?campaign=wolearlyview},
doi = {10.1002/admi.201500755},
year = {2016},
date = {2016-03-17},
journal = {Advanced Materials Interfaces},
volume = {2016},
pages = {1500755},
abstract = {3D interconnected carbon nanotubes (CNTs) are synthesized using an industrially scalable spark plasma technique. At high electric field and elevated temperature under sufficient stress the nanotubes are welded together to form a solid block. The detailed spectroscopic and microscopic analyses show successful welding of the CNTs and formation of interconnected networks. The mechanical characteristics of the 3D CNT block show a high stiffness and yield strength. A full atomistic molecular dynamics simulation elucidates the CNT welding mechanism.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Leonardo Dantas Machado José Moreira de Sousa, Cristiano Francisco Woellner; Galvao, Douglas S.
Carbon Nanoscrolls at High Impacts: A Molecular Dynamics Investigation Journal Article
In: MRS Advances, vol. 2016, 2016.
@article{deSousa2016b,
title = {Carbon Nanoscrolls at High Impacts: A Molecular Dynamics Investigation},
author = {José Moreira de Sousa, Leonardo Dantas Machado, Cristiano Francisco Woellner, Pedro Alves da Silva Autreto and Douglas S. Galvao},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=10242265&fulltextType=RA&fileId=S2059852116002000},
doi = {10.1557/adv.2016.200},
year = {2016},
date = {2016-03-01},
journal = {MRS Advances},
volume = {2016},
abstract = {The behavior of nanostructures under high strain-rate conditions has been object of interest in recent years. For instance, recent experimental investigations showed that at high velocity impacts carbon nanotubes can unzip resulting into graphene nanoribbons. Carbon nanoscrolls (CNS) are among the structures whose high impact behavior has not yet been investigated. CNS are graphene membranes rolled up into papyrus-like structures. Their unique open-ended topology leads to properties not found in close-ended structures, such as nanotubes. Here we report a fully atomistic reactive molecular dynamics study on the behavior of CNS colliding at high velocities against solid targets. Our results show that the velocity and scroll axis orientation are key parameters to determine the resulting formed nanostructures after impact. The relative orientation of the scroll open ends and the substrate is also very important. We observed that for appropriate velocities and orientations, the nanoscrolls can experience large structural deformations and large-scale fractures. We have also observed unscrolling (scrolls going back to planar or quasi-planar graphene membranes), unzip resulting into nanoribbons, and significant reconstructions from breaking and/or formation of new chemical bonds. Another interesting result was that if the CNS impact the substrate with their open ends, for certain velocities, fused scroll walls were observed.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Ygor M. Jaques, Gustavo Brunetto; Galvão, Douglas S.
Nanodroplets Impacting on Graphene Journal Article
In: MRS Advances, vol. 2016, 2016.
@article{Jaques2016b,
title = {Nanodroplets Impacting on Graphene},
author = {Ygor M. Jaques, Gustavo Brunetto and Douglas S. Galvão},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=10253580&fulltextType=RA&fileId=S2059852116002218},
doi = {DOI: 10.1557/adv.2016.221},
year = {2016},
date = {2016-03-01},
journal = {MRS Advances},
volume = {2016},
abstract = {The unique and remarkable properties of graphene can be exploited as the basis to a wide
range of applications. However, in spite of years of investigations there are some important
graphene properties that are not still fully understood, as for example, its wettability. There are
controversial reported results whether graphene is really hydrophobic or hydrophilic. In order to
address this problem we have carried out classical molecular dynamics simulations of water
nanodroplets shot against graphene surface. Our results show that the contact angle values
between the nanodroplets and graphene surfaces depend on the initial droplet velocity value and
these angles can change from 86º (hydrophobic) to 35º (hydrophilic). Our preliminary results
indicate that the graphene wettability can be dependent on spreading liquid dynamics and which
can explain some of the apparent inconsistencies reported in the literature.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
range of applications. However, in spite of years of investigations there are some important
graphene properties that are not still fully understood, as for example, its wettability. There are
controversial reported results whether graphene is really hydrophobic or hydrophilic. In order to
address this problem we have carried out classical molecular dynamics simulations of water
nanodroplets shot against graphene surface. Our results show that the contact angle values
between the nanodroplets and graphene surfaces depend on the initial droplet velocity value and
these angles can change from 86º (hydrophobic) to 35º (hydrophilic). Our preliminary results
indicate that the graphene wettability can be dependent on spreading liquid dynamics and which
can explain some of the apparent inconsistencies reported in the literature.
Pedro Alves da Silva Autreto Cristiano Francisco Woellner, Douglas S. Galvao
One Side-Graphene Hydrogenation (Graphone): Substrate Effects Journal Article
In: MRS Advances, vol. 2016, 2016.
@article{Woellner2016b,
title = {One Side-Graphene Hydrogenation (Graphone): Substrate Effects},
author = {Cristiano Francisco Woellner, Pedro Alves da Silva Autreto, Douglas S. Galvao},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=10234793&fulltextType=RA&fileId=S2059852116001961},
doi = {DOI: 10.1557/adv.2016.196},
year = {2016},
date = {2016-03-01},
journal = {MRS Advances},
volume = {2016},
abstract = {Recent studies on graphene hydrogenation processes showed that hydrogenation occurs via island growing domains, however how the substrate can affect the hydrogenation dynamics and/or pattern formation has not been yet properly investigated. In this work we have addressed these issues through fully atomistic reactive molecular dynamics simulations. We investigated the structural and dynamical aspects of the hydrogenation of graphene membranes (one-side hydrogenation, the so called graphone structure) on different substrates (graphene, few-layers graphene, graphite and platinum). Our results also show that the observed hydrogenation rates are very sensitive to the substrate type. For all investigated cases, the largest fraction of hydrogenated carbon atoms was for platinum substrates. Our results also show that a significant number of randomly distributed H clusters are formed during the early stages of the hydrogenation process, regardless of the type of substrate. These results suggest that, similarly to graphane formation, large perfect graphone-like domains are unlikely to be formed. These findings are especially important since experiments have showed that cluster formation influences the electronic transport properties in hydrogenated graphene.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Daff, Thomas D; Collins, Sean P; Durekova, Hana; Perim, E; Skaf, Munir S; Galvão, Douglas S; Woo, Tom K
Evaluation of carbon nanoscroll materials for post-combustion CO2 capture Journal Article
In: Carbon, vol. 101, pp. 218–225, 2016.
@article{Daff2016,
title = {Evaluation of carbon nanoscroll materials for post-combustion CO2 capture},
author = {Daff, Thomas D and Collins, Sean P and Durekova, Hana and Perim, E and Skaf, Munir S and Galvão, Douglas S and Woo, Tom K},
url = {http://www.sciencedirect.com/science/article/pii/S0008622316300604},
doi = {10.1016/j.carbon.2016.01.072},
year = {2016},
date = {2016-02-11},
journal = {Carbon},
volume = {101},
pages = {218–225},
abstract = {Carbon nanoscrolls are similar to multi-walled carbon nanotubes but constructed from rolled graphene sheets into papyrus-like structures. In this work, molecular simulations are used to evaluate the post-combustion CO2 capture properties of nanoscrolls made of graphene, α-, β-, and γ-graphyne, boron nitride, and three types of carbon nitride. The CO2 uptake capacity, CO2/N2 selectivity and CO2 working capacity were computed with grand canonical Monte Carlo simulations at conditions relevant to post-combustion CO2 capture. The interlayer spacing of the nanoscrolls was optimized for each property and sheet material. For graphene nanoscrolls, the optimal interlayer spacing of 7.3 Å was identified for both the CO2 uptake and selectivity, while for working capacity the optimal interlayer spacing was determined to be 8.6 Å. It was found that the CO2 uptake capacity of the materials correlated to the density of the sheets from which they were formed. Nanoscrolls made from graphene and boron nitride, which have the highest number of atoms per unit area, also showed the highest CO2 uptakes. At 0.15 bar CO2, 313 K, graphene and boron nitride nanoscrolls exhibited exceptional CO2 uptake capacities of 7.7 and 8.2 mmol/g, respectively, while also exhibiting high CO2/N2 selectivities of 135 and 153, respectively. Molecular dynamics simulations were used to examine the adsorption kinetics. The simulations showed that an empty graphene nanoscroll with a roll length of 200 Å could adsorb CO2 into the center of the roll within 10 ns. Materials with pores that can allow CO2 to pass through, such as graphynes, showed much faster adsorption times.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Jaques, Ygor M.; Brunetto, Gustavo; Galvao, Douglas S.
Nanodroplets Impacting on Graphene Online
2016, ((ArXiv preprint)).
@online{Jaques2016,
title = {Nanodroplets Impacting on Graphene},
author = {Jaques, Ygor M. and Brunetto, Gustavo and Galvao, Douglas S.},
url = {http://arxiv.org/abs/1602.02013},
year = {2016},
date = {2016-02-05},
abstract = {The unique and remarkable properties of graphene can be exploited as the basis to a wide
range of applications. However, in spite of years of investigations there are some important
graphene properties that are not still fully understood, as for example, its wettability. There are
controversial reported results whether graphene is really hydrophobic or hydrophilic. In order to
address this problem we have carried out classical molecular dynamics simulations of water
nanodroplets shot against graphene surface. Our results show that the contact angle values
between the nanodroplets and graphene surfaces depend on the initial droplet velocity value and
these angles can change from 86º (hydrophobic) to 35º (hydrophilic). Our preliminary results
indicate that the graphene wettability can be dependent on spreading liquid dynamics and which
can explain some of the apparent inconsistencies reported in the literature.},
note = {(ArXiv preprint)},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
range of applications. However, in spite of years of investigations there are some important
graphene properties that are not still fully understood, as for example, its wettability. There are
controversial reported results whether graphene is really hydrophobic or hydrophilic. In order to
address this problem we have carried out classical molecular dynamics simulations of water
nanodroplets shot against graphene surface. Our results show that the contact angle values
between the nanodroplets and graphene surfaces depend on the initial droplet velocity value and
these angles can change from 86º (hydrophobic) to 35º (hydrophilic). Our preliminary results
indicate that the graphene wettability can be dependent on spreading liquid dynamics and which
can explain some of the apparent inconsistencies reported in the literature.
Xifan Wang Sidong Lei, Bo Li
Surface functionalization of two-dimensional metal chalcogenides by Lewis acid–base chemistry Journal Article
In: Nature Nanotechnology, vol. 11, pp. 465–471, 2016.
@article{Lei2016,
title = {Surface functionalization of two-dimensional metal chalcogenides by Lewis acid–base chemistry},
author = {Sidong Lei, Xifan Wang, Bo Li, Jiahao Kang, Yongmin He, Antony George, Liehui Ge, Yongji Gong, Pei Dong, Zehua Jin, Gustavo Brunetto, Weibing Chen, Zuan-Tao Lin, Robert Baines, Douglas S. Galvão, Jun Lou, Enrique Barrera, Kaustav Banerjee, Robert Vajtai & Pulickel Ajayan},
url = {http://www.nature.com/nnano/journal/vaop/ncurrent/full/nnano.2015.323.html},
doi = {10.1038/nnano.2015.323},
year = {2016},
date = {2016-02-01},
journal = {Nature Nanotechnology},
volume = {11},
pages = {465–471},
abstract = {Precise control of the electronic surface states of two-dimensional (2D) materials could improve their versatility and widen their applicability in electronics and sensing. To this end, chemical surface functionalization has been used to adjust the electronic properties of 2D materials. So far, however, chemical functionalization has relied on lattice defects and physisorption methods that inevitably modify the topological characteristics of the atomic layers. Here we make use of the lone pair electrons found in most of 2D metal chalcogenides and report a functionalization method via a Lewis acid–base reaction that does not alter the host structure. Atomic layers of n-type InSe react with Ti4+ to form planar p-type [Ti4+n(InSe)] coordination complexes. Using this strategy, we fabricate planar p–n junctions on 2D InSe with improved rectification and photovoltaic properties, without requiring heterostructure growth procedures or device fabrication processes. We also show that this functionalization approach works with other Lewis acids (such as B3+, Al3+ and Sn4+) and can be applied to other 2D materials (for example MoS2, MoSe2). Finally, we show that it is possible to use Lewis acid–base chemistry as a bridge to connect molecules to 2D atomic layers and fabricate a proof-of-principle dye-sensitized photosensing device.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
de Sousa, Jose Moreira; Machado, Leonardo Dantas; Woellner, Cristiano Francisco; Autreto, Pedro Alves da Silva; Galvao, Douglas S
Carbon Nanoscrolls at High Impacts: A Molecular Dynamics Investigation Online
2016, ((ArXiv Preprint)).
@online{deSousa2016b,
title = {Carbon Nanoscrolls at High Impacts: A Molecular Dynamics Investigation},
author = {de Sousa, Jose Moreira and Machado, Leonardo Dantas and Woellner, Cristiano Francisco and Autreto, Pedro Alves da Silva and Galvao, Douglas S},
url = {http://arxiv.org/abs/1601.04875},
year = {2016},
date = {2016-01-19},
abstract = {The behavior of nanostructures under high strain-rate conditions has been object of interest in recent years. For instance, recent experimental investigations showed that at high velocity impacts carbon nanotubes can unzip resulting into graphene nanoribbons. Carbon nanoscrolls (CNS) are among the structures whose high impact behavior has not yet been investigated. CNS are graphene membranes rolled up into papyrus-like structures. Their unique open-ended topology leads to properties not found in close-ended structures, such as nanotubes. Here we report a fully atomistic reactive molecular dynamics study on the behavior of CNS colliding at high velocities against solid targets. Our results show that the velocity and scroll axis orientation are key parameters to determine the resulting formed nanostructures after impact. The relative orientation of the scroll open ends and the substrate is also very important. We observed that for appropriate velocities and orientations, the nanoscrolls can experience large structural deformations and large-scale fractures. We have also observed unscrolling (scrolls going back to planar or quasi-planar graphene membranes), unzip resulting into nanoribbons, and significant reconstructions from breaking and/or formation of new chemical bonds. Another interesting result was that if the CNS impact the substrate with their open ends, for certain velocities, fused scroll walls were observed.},
note = {(ArXiv Preprint)},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Woellner, Cristiano Francisco; Autreto, Pedro Alves da Silva; Galvao, Douglas S
One Side-Graphene Hydrogenation (Graphone): Substrate Effects Online
2016, visited: 18.01.2016, ((ArXiv preprint)).
@online{Woellner2016,
title = {One Side-Graphene Hydrogenation (Graphone): Substrate Effects},
author = {Woellner, Cristiano Francisco and Autreto, Pedro Alves da Silva and Galvao, Douglas S},
url = {http://arxiv.org/abs/1601.04484},
year = {2016},
date = {2016-01-18},
urldate = {2016-01-18},
abstract = {Recent studies on graphene hydrogenation processes showed that hydrogenation occurs
via island growing domains, however how the substrate can affect the hydrogenation dynamics
and/or pattern formation has not been yet properly investigated. In this work we have addressed
these issues through fully atomistic reactive molecular dynamics simulations. We investigated
the structural and dynamical aspects of the hydrogenation of graphene membranes (one-side
hydrogenation, the so called graphone structure) on different substrates (graphene, few-layers
graphene, graphite and platinum). Our results also show that the observed hydrogenation rates
are very sensitive to the substrate type. For all investigated cases, the largest fraction of
hydrogenated carbon atoms was for platinum substrates. Our results also show that a significant
number of randomly distributed H clusters are formed during the early stages of the
hydrogenation process, regardless of the type of substrate and temperature. These results suggest
that, similarly to graphane formation, large perfect graphone-like domains are unlikely to be
formed. These findings are especially important since experiments have showed that cluster
formation influences the electronic transport properties in hydrogenated graphene.},
note = {(ArXiv preprint)},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
via island growing domains, however how the substrate can affect the hydrogenation dynamics
and/or pattern formation has not been yet properly investigated. In this work we have addressed
these issues through fully atomistic reactive molecular dynamics simulations. We investigated
the structural and dynamical aspects of the hydrogenation of graphene membranes (one-side
hydrogenation, the so called graphone structure) on different substrates (graphene, few-layers
graphene, graphite and platinum). Our results also show that the observed hydrogenation rates
are very sensitive to the substrate type. For all investigated cases, the largest fraction of
hydrogenated carbon atoms was for platinum substrates. Our results also show that a significant
number of randomly distributed H clusters are formed during the early stages of the
hydrogenation process, regardless of the type of substrate and temperature. These results suggest
that, similarly to graphane formation, large perfect graphone-like domains are unlikely to be
formed. These findings are especially important since experiments have showed that cluster
formation influences the electronic transport properties in hydrogenated graphene.
Vinod, Soumya; Tiwary, Chandra Sekhar; Machado, Leonardo Dantas; Ozden, Sehmus; Shaw, Preston; Cho, Juny; Vajtai, Robert; Galvao, Douglas Soares; Ajayan, Pulickel M
Strain Rate Dependent Shear Plasticity in Graphite Oxide Journal Article
In: Nano Letters, vol. 16, no. 2, pp. 1127–1131, 2016.
@article{Vinod2016,
title = {Strain Rate Dependent Shear Plasticity in Graphite Oxide},
author = {Vinod, Soumya and Tiwary, Chandra Sekhar and Machado, Leonardo Dantas and Ozden, Sehmus and Shaw, Preston and Cho, Juny and Vajtai, Robert and Galvao, Douglas Soares and Ajayan, Pulickel M},
url = {http://pubs.acs.org/doi/abs/10.1021/acs.nanolett.5b04346},
doi = {10.1021/acs.nanolett.5b04346},
year = {2016},
date = {2016-01-16},
journal = {Nano Letters},
volume = {16},
number = {2},
pages = {1127–1131},
abstract = {Graphene oxide film is made of stacked graphene layers with chemical functionalities, and we report that plasticity in the film can be engineered by strain rate tuning. The deformation behavior and plasticity of such functionalized layered systems is dominated by shear slip between individual layers and interaction between functional groups. Stress–strain behavior and theoretical models suggest that the deformation is strongly strain rate dependent and undergoes brittle to ductile transition with decreasing strain rate.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
G. Brunetto J.M. de Sousa, V. R. Coluci
Torsional “superplasticity” of graphyne nanotubes Journal Article
In: Carbon, vol. 96, pp. 14-19, 2016.
@article{deSousa2016,
title = {Torsional “superplasticity” of graphyne nanotubes},
author = {J.M. de Sousa, G. Brunetto, V.R. Coluci, D.S. Galvao },
url = {http://www.sciencedirect.com/science/article/pii/S000862231530258X},
doi = { http://dx.doi.org/10.1016/j.carbon.2015.09.039},
year = {2016},
date = {2016-01-01},
journal = {Carbon},
volume = {96},
pages = {14-19},
abstract = {Graphyne is a planar two-dimensional carbon allotrope formed by atoms in sp, sp2, and sp3 hybridized states. Topologically graphyne nanotubes (GNTs) can be considered as cylindrically rolled up graphyne sheets, similarly as carbon nanotubes (CNTs) can be considered rolled up graphene sheets. Due to the presence of single, double, and triple bonds, GNTs exhibit porous sidewalls that can be exploited in many diverse applications. In this work, we investigated the mechanical behavior of GNTs under torsional strains through reactive molecular dynamics simulations. Our results show that GNTs are more flexible than CNTs and exhibit “superplasticit”, with fracture angles that are up to 35 times higher than the ones reported to CNTs. This GNT “superplastic” behavior can be explained in terms of irreversible recon- struction processes (mainly associated with the triple bonds) that occur during torsional strains.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Dong, Pei; Chipara, Alin Cristian; Loya, Phillip; Yang, Yingchao; Ge, Liehui; Lei, Sidong; Li, Bo; Brunetto, Gustavo; Machado, Leonardo Dantas; Hong, Liang; others,
A Solid-liquid Self-adaptive Polymeric Composite Journal Article
In: ACS Applied Materials & Interfaces, vol. 8, no. 3, pp. 2142–2147, 2016.
@article{Dong2016,
title = {A Solid-liquid Self-adaptive Polymeric Composite},
author = {Dong, Pei and Chipara, Alin Cristian and Loya, Phillip and Yang, Yingchao and Ge, Liehui and Lei, Sidong and Li, Bo and Brunetto, Gustavo and Machado, Leonardo Dantas and Hong, Liang and others},
url = {http://pubs.acs.org/doi/abs/10.1021/acsami.5b10667},
doi = {10.1021/acsami.5b10667},
year = {2016},
date = {2016-01-01},
journal = {ACS Applied Materials & Interfaces},
volume = {8},
number = {3},
pages = {2142–2147},
abstract = {A solid–liquid self-adaptive composite (SAC) is synthesized using a simple mixing–evaporation protocol, with poly(dimethylsiloxane) (PDMS) and poly(vinylidene fluoride) (PVDF) as active constituents. SAC exists as a porous solid containing a near equivalent distribution of the solid (PVDF)–liquid (PDMS) phases, with the liquid encapsulated and stabilized within a continuous solid network percolating throughout the structure. The pores, liquid, and solid phases form a complex hierarchical structure, which offers both mechanical robustness and a significant structural adaptability under external forces. SAC exhibits attractive self-healing properties during tension, and demonstrates reversible self-stiffening properties under compression with a maximum of 7-fold increase seen in the storage modulus. In a comparison to existing self-healing and self-stiffening materials, SAC offers distinct advantages in the ease of fabrication, high achievable storage modulus, and reversibility. Such materials could provide a new class of adaptive materials system with multifunctionality, tunability, and scale-up potentials.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
de Sousa, Jose M.; Autreto, Pedro A. S.; Galvao, Douglas S.
Hydrogenation Dynamics of Twisted Carbon Nanotubes Online
2015, (ArXiv preprint).
@online{deSousa2015,
title = {Hydrogenation Dynamics of Twisted Carbon Nanotubes},
author = {Jose M. de Sousa and Pedro A. S. Autreto and Douglas S. Galvao},
url = {http://arxiv.org/abs/1510.00265},
year = {2015},
date = {2015-10-01},
abstract = {Carbon Nanotubes (CNTs) are one of the most important materials in nanotechnology. In some of their technological applications (electromechanical oscillators and mechanical actuators for artificial muscles, for instance), it is necessary to subject them to large deformations. Although this frequently happens in air, there are only few studies about the interaction of deformed CNTs with the atmosphere and the dynamics of these processes has not yet been addressed. In this work, we have investigated, through fully atomistic reactive molecular dynamics simulations, the process of hydrogenation of highly twisted CNTs. Our results show that hydrogenation effective ratio is directly related to the tube twist angle values and can lead to twisted tube fractures with well defined patterns (unzip-like). Our results also show that these fracture processes can be exploited to controllably produce graphene nanoribbons.},
note = {ArXiv preprint},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Gustavo Brunetto Jose M. de Sousa, Vitor R. Coluci
Torsional "Superplasticity" of Graphyne Nanotubes Online
2015, (ArXiv reprint of Torsional "Superplasticity" of Graphyne Nanotubes, published in Carbon 96, 14 (2016).).
@online{deSousa2015b,
title = {Torsional "Superplasticity" of Graphyne Nanotubes},
author = {Jose M. de Sousa, Gustavo Brunetto, Vitor R. Coluci, Douglas S. Galvao},
url = {http://arxiv.org/abs/1509.08746},
year = {2015},
date = {2015-09-29},
abstract = {Graphyne is a planar two-dimensional carbon allotrope formed by atoms in sp, sp2, and sp3 hybridized states. Topologically graphyne nanotubes (GNTs) can be considered as cylindrically rolled up graphyne sheets, similarly as carbon nanotubes (CNTs) can be considered rolled up graphene sheets. Due to the presence of single, double, and triple bonds, GNTs exhibit porous sidewalls that can be exploited in many diverse applications. In this work, we investigated the mechanical behavior of GNTs under torsional strains through reactive molecular dynamics simulations. Our results show that GNTs are more flexible than CNTs and exhibit 'superplasticity', with fracture angles that are up to 35 times higher than the ones reported to CNTs. This GNT 'superplastic' behavior can be explained in terms of irreversible reconstruction processes (mainly associated with the triple bonds) that occur during torsional strains.},
note = {ArXiv reprint of Torsional "Superplasticity" of Graphyne Nanotubes, published in Carbon 96, 14 (2016).},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
S Fang ZF Liu, FA Moura
Hierarchically buckled sheath-core fibers for superelastic electronics, sensors, and muscles Journal Article
In: Science, vol. 349, no. 6246, pp. 404-404, 2015.
@article{Liu2015,
title = {Hierarchically buckled sheath-core fibers for superelastic electronics, sensors, and muscles},
author = {ZF Liu, S Fang, FA Moura, JN Ding, N Jiang, J Di, M Zhang, X Lepró, DS Galvão, CS Haines, NY Yuan, SG Yin, DW Lee, R Wang, HY Wang, W Lv, C Dong, RC Zhang, MJ Chen, Q Yin, YT Chong, R Zhang, X Wang, MD Lima, R Ovalle-Robles, D Qian, H Lu, RH Baughman},
url = {http://www.sciencemag.org/content/349/6246/400.full.pdf},
doi = {10.1126/science.aaa7952},
year = {2015},
date = {2015-07-24},
journal = {Science},
volume = {349},
number = {6246},
pages = {404-404},
abstract = {Superelastic conducting fibers with improved properties and functionalities are needed
for diverse applications. Here we report the fabrication of highly stretchable (up to 1320%)
sheath-core conducting fibers created by wrapping carbon nanotube sheets oriented in
the fiber direction on stretched rubber fiber cores. The resulting structure exhibited
distinct short- and long-period sheath buckling that occurred reversibly out of phase
in the axial and belt directions, enabling a resistance change of less than 5% for a
1000% stretch. By including other rubber and carbon nanotube sheath layers, we
demonstrated strain sensors generating an 860% capacitance change and electrically
powered torsional muscles operating reversibly by a coupled tension-to-torsion
actuation mechanism. Using theory, we quantitatively explain the complementary effects
of an increase in muscle length and a large positive Poisson’s ratio on torsional actuation
and electronic properties.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
for diverse applications. Here we report the fabrication of highly stretchable (up to 1320%)
sheath-core conducting fibers created by wrapping carbon nanotube sheets oriented in
the fiber direction on stretched rubber fiber cores. The resulting structure exhibited
distinct short- and long-period sheath buckling that occurred reversibly out of phase
in the axial and belt directions, enabling a resistance change of less than 5% for a
1000% stretch. By including other rubber and carbon nanotube sheath layers, we
demonstrated strain sensors generating an 860% capacitance change and electrically
powered torsional muscles operating reversibly by a coupled tension-to-torsion
actuation mechanism. Using theory, we quantitatively explain the complementary effects
of an increase in muscle length and a large positive Poisson’s ratio on torsional actuation
and electronic properties.
Chandra Sekhar Tiwary Dibyendu Chakravarty, Leonardo Dantas Machado
Zirconia-Nanoparticle-Reinforced Morphology-Engineered Graphene-Based Foams Journal Article
In: Advanced Materials, vol. 27, no. 31, pp. 4534–4543, 2015.
@article{Chakravarty2015,
title = {Zirconia-Nanoparticle-Reinforced Morphology-Engineered Graphene-Based Foams},
author = { Dibyendu Chakravarty , Chandra Sekhar Tiwary , Leonardo Dantas Machado ,
Gustavo Brunetto , Soumya Vinod , Ram Manohar Yadav , Douglas S. Galvao ,
Shrikant V. Joshi , Govindan Sundararajan, Pulickel M. Ajayan },
url = {http://onlinelibrary.wiley.com/doi/10.1002/adma.201502409/full},
doi = {10.1002/adma.201502409},
year = {2015},
date = {2015-07-15},
journal = {Advanced Materials},
volume = {27},
number = {31},
pages = {4534–4543},
abstract = {The morphology of graphene-based foams can be engineered by reinforcing them with nanocrystalline zirconia, thus improving their oil-adsorption capacity; This can be observed experimentally and explained theoretically. Low zirconia fractions yield flaky microstructures where zirconia nanoparticles arrest propagating cracks. Higher zirconia concentrations possess a mesh-like interconnected structure where the degree of coiling is dependant on the local zirconia content.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Yongji Gong Kunttal Keyshar, Gonglan Ye
Chemical Vapor Deposition of Monolayer Rhenium Disulfide (ReS2) Journal Article
In: Advanced Materials, vol. 27, no. 31, pp. 4640–4648, 2015.
@article{Keyshar2015,
title = {Chemical Vapor Deposition of Monolayer Rhenium Disulfide (ReS2)},
author = {Kunttal Keyshar , Yongji Gong , Gonglan Ye , Gustavo Brunetto , Wu Zhou ,
Daniel P. Cole , Ken Hackenberg , Yongmin He , Leonardo Machado , Mohamad Kabbani ,
Amelia H. C. Hart , Bo Li , Douglas S. Galvao , Antony George , Robert Vajtai ,
Chandra Sekhar Tiwary , Pulickel M. Ajayan},
url = {http://onlinelibrary.wiley.com/doi/10.1002/adma.201501795/full},
doi = {10.1002/adma.201501795},
year = {2015},
date = {2015-07-03},
journal = {Advanced Materials},
volume = {27},
number = {31},
pages = {4640–4648},
abstract = {The direct synthesis of monolayer and multilayer ReS2 by chemical vapor deposition at a low temperature of 450 °C is reported. Detailed characterization of this material is performed using various spectroscopy and microscopy methods. Furthermore initial field-effect transistor characteristics are evaluated, which highlight the potential in being used as an n-type semiconductor.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Andrei V Alaferdov Victor A Ermakov, Alfredo R Vaz
Burning Graphene Layer-by-Layer Journal Article
In: Nature Scientific Reports, vol. 5, pp. 11546, 2015.
@article{Ermakov2015,
title = {Burning Graphene Layer-by-Layer},
author = {Victor A Ermakov, Andrei V Alaferdov, Alfredo R Vaz, Eric Perim, Pedro AS Autreto, Ricardo Paupitz, Douglas S Galvao, Stanislav A Moshkalev},
url = {http://www.nature.com/articles/srep11546?WT.ec_id=SREP-639-20150630},
doi = {10.1038/srep11546},
year = {2015},
date = {2015-06-23},
journal = {Nature Scientific Reports},
volume = {5},
pages = {11546},
abstract = {Graphene, in single layer or multi-layer forms, holds great promise for future electronics and high-temperature applications. Resistance to oxidation, an important property for high-temperature applications, has not yet been extensively investigated. Controlled thinning of multi-layer graphene (MLG), e.g., by plasma or laser processing is another challenge, since the existing methods produce non-uniform thinning or introduce undesirable defects in the basal plane. We report here that heating to extremely high temperatures (exceeding 2000 K) and controllable layer-by-layer burning (thinning) can be achieved by low-power laser processing of suspended high-quality MLG in air in “cold-wall” reactor configuration. In contrast, localized laser heating of supported samples results in non-uniform graphene burning at much higher rates. Fully atomistic molecular dynamics simulations were also performed to reveal details of oxidation mechanisms leading to uniform layer-by-layer graphene gasification. The extraordinary resistance of MLG to oxidation paves the way to novel high-temperature applications as continuum light source or scaffolding material.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Wesller G Schmidt Abraham G Cano-Marquez, Jenaina Ribeiro-Soares
Enhanced Mechanical Stability of Gold Nanotips through Carbon Nanocone Encapsulation Journal Article
In: Nature Scientific Reports, vol. 5, pp. 10408, 2015.
@article{Cano-Marquez2015,
title = {Enhanced Mechanical Stability of Gold Nanotips through Carbon Nanocone Encapsulation},
author = {Abraham G Cano-Marquez, Wesller G Schmidt, Jenaina Ribeiro-Soares, Luiz Gustavo Cançado, Wagner N Rodrigues, Adelina P Santos, Clascidia A Furtado, Pedro AS Autreto, Ricardo Paupitz, Douglas S Galvão, Ado Jorio},
url = {http://www.nature.com/articles/srep10408},
doi = {10.1038/srep10408},
year = {2015},
date = {2015-06-17},
journal = {Nature Scientific Reports},
volume = {5},
pages = {10408},
abstract = {Gold is a noble metal that, in comparison with silver and copper, has the advantage of corrosion resistance. Despite its high conductivity, chemical stability and biocompatibility, gold exhibits high plasticity, which limits its applications in some nanodevices. Here, we report an experimental and theoretical study on how to attain enhanced mechanical stability of gold nanotips. The gold tips were fabricated by chemical etching and further encapsulated with carbon nanocones via nanomanipulation. Atomic force microscopy experiments were carried out to test their mechanical stability. Molecular dynamics simulations show that the encapsulated nanocone changes the strain release mechanisms at the nanoscale by blocking gold atomic sliding, redistributing the strain along the whole nanostructure. The carbon nanocones are conducting and can induce magnetism, thus opening new avenues on the exploitation of transport, mechanical and magnetic properties of gold covered by sp2 carbon at the nanoscale.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2005
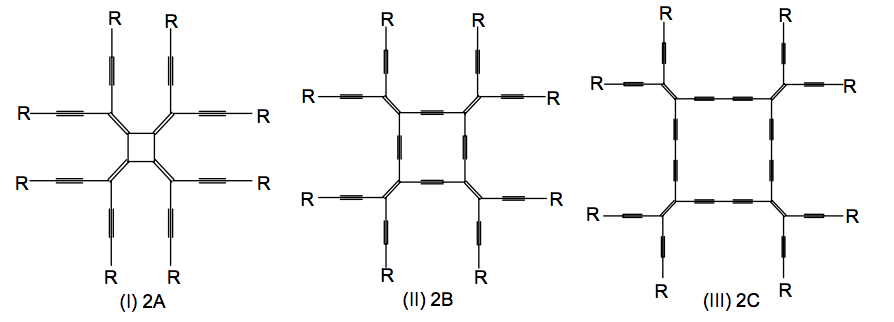
Konstantinova, Elena; Galvao, Douglas S; Barone, Paulo MVB; Dantas, Socrates O
Structural and electronic properties of radialenes and related systems Journal Article
In: Journal of Molecular Structure: THEOCHEM, vol. 729, no. 3, pp. 203–210, 2005.
Abstract | Links | BibTeX | Tags: DFT, Electronic Structure, Radialenes
@article{konstantinova2005structural,
title = {Structural and electronic properties of radialenes and related systems},
author = {Konstantinova, Elena and Galvao, Douglas S and Barone, Paulo MVB and Dantas, Socrates O},
url = {http://www.sciencedirect.com/science/article/pii/S016612800500480X},
year = {2005},
date = {2005-01-01},
journal = {Journal of Molecular Structure: THEOCHEM},
volume = {729},
number = {3},
pages = {203--210},
publisher = {Elsevier},
abstract = {The discovery of new allotropic forms of carbon gives rise to a great interest in carbon compounds as building blocks for novel nanostructure materials. Radialenes are homologous series of compounds with a cycloalkane nucleus bound to methylene side groups, with molecular formula CnHn. The series of expanded radialenes of molecular formulae C2nHn and C3nHn are obtained by inserting acetylene or diacetylene groups between each pair of methylene units. This paper is a report on the theoretical study of structural, electronic and spectroscopic properties of radialenes, expanded radialenes and related molecular systems. Using semiempirical methods we explore the behavior of π-electrons along the carbon-rich skeleton. The results for structural parameters are in a good agreement with the available experimental data. The calculated electronic gaps and spatial distribution of frontier orbitals indicate to interesting electrical and nonlinear optical properties of the explored compounds, which may be useful for technological applications.},
keywords = {DFT, Electronic Structure, Radialenes},
pubstate = {published},
tppubtype = {article}
}

Troche, Karla S; Braga, Scheila F; Coluci, Vitor R; Galvao, Douglas S
Carcinogenic classification of polycyclic aromatic hydrocarbons through theoretical descriptors Journal Article
In: International journal of quantum chemistry, vol. 103, no. 5, pp. 718–730, 2005.
Abstract | Links | BibTeX | Tags: Carcinogenesis, HCA, Neural Networks, PCA, Polycyclic Aromatic Hydrocarbons (PAHs), Theory of Electronic Indices
@article{troche2005carcinogenic,
title = {Carcinogenic classification of polycyclic aromatic hydrocarbons through theoretical descriptors},
author = {Troche, Karla S and Braga, Scheila F and Coluci, Vitor R and Galvao, Douglas S},
url = {http://onlinelibrary.wiley.com/doi/10.1002/qua.20529/full},
year = {2005},
date = {2005-01-01},
journal = {International journal of quantum chemistry},
volume = {103},
number = {5},
pages = {718--730},
publisher = {Wiley Online Library},
abstract = {Polycyclic aromatic hydrocarbons (PAHs) constitute an importantfamily of molecules capable of inducing chemical carcinogenesis. In this work we reporta comparative structure–activity relationship (SAR) study for 81 PAHs using differentmethodologies. The recently developed electronic indices methodology (EIM) withquantum descriptors obtained from different semiempirical methods (AM1, PM3, andPM5) was contrasted against more standard pattern recognition methods (PRMs),principal component analysis (PCA), hierarchical cluster analysis (HCA), Kth nearestneighbor (KNN), soft independent modeling of class analogies (SIMCA), and neuralnetworks (NN). Our results show that PRMs validate the statistical value of electronicparameters derived from EIM analysis and their ability to identify active compounds.EIM outperformed more standard SAR methodologies and does not appear to besignificantly Hamiltonian-dependent.},
keywords = {Carcinogenesis, HCA, Neural Networks, PCA, Polycyclic Aromatic Hydrocarbons (PAHs), Theory of Electronic Indices},
pubstate = {published},
tppubtype = {article}
}
Campos, Paulo RA; de Oliveira, Viviane M; Giro, Ronaldo; Galvao, Douglas S
Campos et al. Reply Journal Article
In: Physical Review Letters, vol. 95, no. 22, pp. 229802, 2005.
Links | BibTeX | Tags: Cellular Automata, Cicadas, Prime Numbers
@article{campos2005campos,
title = {Campos et al. Reply},
author = {Campos, Paulo RA and de Oliveira, Viviane M and Giro, Ronaldo and Galvao, Douglas S},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.95.229802},
year = {2005},
date = {2005-01-01},
journal = {Physical Review Letters},
volume = {95},
number = {22},
pages = {229802},
publisher = {APS},
keywords = {Cellular Automata, Cicadas, Prime Numbers},
pubstate = {published},
tppubtype = {article}
}
da Fonseca, Alexandre F; Galvao, Douglas S; Malta, Coraci P
Binormal nanohelices Proceedings
Cambridge University Press, vol. 903, 2005.
Abstract | Links | BibTeX | Tags: Elasticity, Filaments, Nanowires
@proceedings{da2005binormal,
title = {Binormal nanohelices},
author = {da Fonseca, Alexandre F and Galvao, Douglas S and Malta, Coraci P},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8108794&fileId=S1946427400047680},
year = {2005},
date = {2005-01-01},
journal = {MRS Proceedings},
volume = {903},
pages = {0903--Z14},
publisher = {Cambridge University Press},
abstract = {Helical structures can be classified in accordance with the orientation of its cross-section with respect to the normal or binormal vectors. We investigate the geometric features of several nanosprings verifying the non-existence of normal nanohelices. In this work, using the VLS growth model, we explain not only the absence of normal nanosprings but also the growing process of binormal nanosprings. The dynamical stability of crystalline ZnO binormal nanohelices is also addressed.},
keywords = {Elasticity, Filaments, Nanowires},
pubstate = {published},
tppubtype = {proceedings}
}

Coluci, Vitor; Braga, Scheila F; Baughman, Ray H; Galvao, Douglas S
Hydrogen Storage in Carbon Nanoscrolls: A Molecular Dynamics Study Proceedings
Cambridge University Press, vol. 885, 2005.
Abstract | Links | BibTeX | Tags: Hydrogen Storage, Molecular Dynamics, Monte Carlo, Scrolls
@proceedings{coluci2005hydrogen,
title = {Hydrogen Storage in Carbon Nanoscrolls: A Molecular Dynamics Study},
author = {Coluci, Vitor and Braga, Scheila F and Baughman, Ray H and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8012272&fileId=S1946427400039816},
year = {2005},
date = {2005-01-01},
journal = {MRS Proceedings},
volume = {885},
pages = {0885--A06},
publisher = {Cambridge University Press},
abstract = {We carried out molecular dynamics simulations with Tersoff-Brenner potentials in order to investigate the hydrogen uptake mechanisms and storage capacity of carbon nanoscrolls (CNSs). CNSs are jelly roll-like structures formed by wrapping graphene layers. Interlayer adsorption is an option for this material, which does not exist for single and multiwalled carbon nanotubes. We analyzed the processes of hydrogen physisorption and uptake mechanisms. We observed incorporation of hydrogen molecules in both external and internal scroll surfaces. Insertion in the internal cavity and between the scroll layers is responsible for 40% of the total hydrogen adsorption at 77 K.},
keywords = {Hydrogen Storage, Molecular Dynamics, Monte Carlo, Scrolls},
pubstate = {published},
tppubtype = {proceedings}
}
2004

Braga, Scheila F; Coluci, Vitor R; Legoas, Sergio B; Giro, Ronaldo; Galvao, Douglas S; Baughman, Ray H
Structure and dynamics of carbon nanoscrolls Journal Article
In: Nano Letters, vol. 4, no. 5, pp. 881–884, 2004.
Abstract | Links | BibTeX | Tags: Molecular Dynamics, Scrolls, Structure
@article{braga2004structure,
title = {Structure and dynamics of carbon nanoscrolls},
author = {Braga, Scheila F and Coluci, Vitor R and Legoas, Sergio B and Giro, Ronaldo and Galvao, Douglas S and Baughman, Ray H},
url = {http://pubs.acs.org/doi/abs/10.1021/nl0497272},
year = {2004},
date = {2004-01-01},
journal = {Nano Letters},
volume = {4},
number = {5},
pages = {881--884},
publisher = {American Chemical Society},
abstract = {Carbon nanotube scrolls (CNSs) provide an interesting form of carbon that ideally consists of a single sheet of graphite that is spiral wrapped
to form a nanotube. We here use molecular dynamics simulations to investigate CNS formation, stability, and the structural effects due to
charge injection. CNS formation is seen to automatically occur when a critical overlap between sheet layers is achieved for the partially curled
sheet. We find that charge injection causes unwinding of the CNSs, which might be important for the application of CNSs as nanomechanical
actuators},
keywords = {Molecular Dynamics, Scrolls, Structure},
pubstate = {published},
tppubtype = {article}
}
to form a nanotube. We here use molecular dynamics simulations to investigate CNS formation, stability, and the structural effects due to
charge injection. CNS formation is seen to automatically occur when a critical overlap between sheet layers is achieved for the partially curled
sheet. We find that charge injection causes unwinding of the CNSs, which might be important for the application of CNSs as nanomechanical
actuators

Legoas, SB; Coluci, VR; Braga, SF; Coura, PZ; Dantas, SO; Galvao, DS
Gigahertz nanomechanical oscillators based on carbon nanotubes Journal Article
In: Nanotechnology, vol. 15, no. 4, pp. S184, 2004.
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, Molecular Dynamics, Oscillators
@article{legoas2004gigahertz,
title = {Gigahertz nanomechanical oscillators based on carbon nanotubes},
author = {Legoas, SB and Coluci, VR and Braga, SF and Coura, PZ and Dantas, SO and Galvao, DS},
url = {http://iopscience.iop.org/0957-4484/15/4/012},
year = {2004},
date = {2004-01-01},
journal = {Nanotechnology},
volume = {15},
number = {4},
pages = {S184},
publisher = {IOP Publishing},
abstract = {We report molecular dynamics studies of carbon nanotubes as mechanical gigahertz oscillators. Our results show that different oscillatory regimes exist but that sustained oscillations are possible only when the radii difference values of the inner and outer tubes are {sim }3.4~AA . Frequencies as large as 87 GHz were obtained. Calculated force and frequency values are in good agreement with estimated data from recent experimental investigations.},
keywords = {Carbon Nanotubes, Molecular Dynamics, Oscillators},
pubstate = {published},
tppubtype = {article}
}
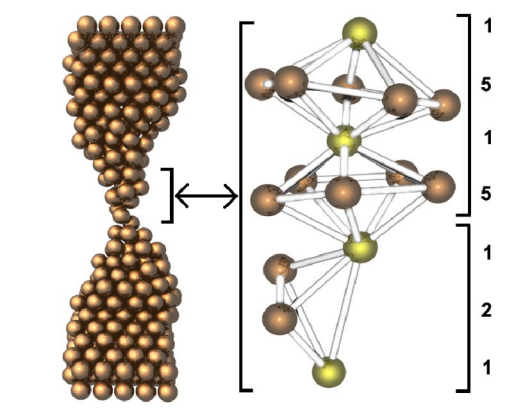
Gonzalez, JC; Rodrigues, V; Bettini, J; Rego, LGC; Rocha, AR; Coura, PZ; Dantas, SO; Sato, F; Galvao, DS; Ugarte, D
Indication of unusual pentagonal structures in atomic-size Cu nanowires Journal Article
In: Physical Review Letters, vol. 93, no. 12, pp. 126103, 2004.
Abstract | Links | BibTeX | Tags: Copper Nanowires, Linear Atomic Chains, Metallic Nanowires, Molecular Dynamics, TEM, top20
@article{gonzalez2004indication,
title = {Indication of unusual pentagonal structures in atomic-size Cu nanowires},
author = {Gonzalez, JC and Rodrigues, V and Bettini, J and Rego, LGC and Rocha, AR and Coura, PZ and Dantas, SO and Sato, F and Galvao, DS and Ugarte, D},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.93.126103},
year = {2004},
date = {2004-01-01},
journal = {Physical Review Letters},
volume = {93},
number = {12},
pages = {126103},
publisher = {APS},
abstract = {We present a study of the structural and quantum conductance properties of atomic-size copper nanowires generated by mechanical stretching. The atomistic evolution was derived from time-resolved electron microscopy observations and molecular dynamics simulations. We have analyzed the quantum transport behavior by means of conductance measurements and theoretical calculations. The results suggest the formation of an unusual and highly stable pentagonal Cu nanowire with a diameter of ∼0.45 nm and ∼4.5 conductance quanta.
},
keywords = {Copper Nanowires, Linear Atomic Chains, Metallic Nanowires, Molecular Dynamics, TEM, top20},
pubstate = {published},
tppubtype = {article}
}
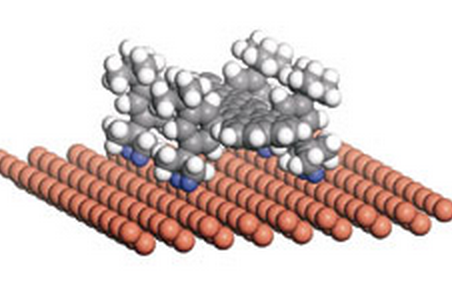
Otero, Roberto; Hummelink, Frauke; Sato, Fernando; Legoas, Sergio B; Thostrup, Peter; Lægsgaard, Erik; Stensgaard, Ivan; Galvao, Douglas S; Besenbacher, Flemming
Lock-and-key effect in the surface diffusion of large organic molecules probed by STM Journal Article
In: Nature Materials, vol. 3, no. 11, pp. 779–782, 2004.
Abstract | Links | BibTeX | Tags: Landers, Molecular Dynamics, Molecular Electronics, STM, top20
@article{otero2004lock,
title = {Lock-and-key effect in the surface diffusion of large organic molecules probed by STM},
author = {Otero, Roberto and Hummelink, Frauke and Sato, Fernando and Legoas, Sergio B and Thostrup, Peter and Lægsgaard, Erik and Stensgaard, Ivan and Galvao, Douglas S and Besenbacher, Flemming},
url = {http://www.nature.com/nmat/journal/v3/n11/full/nmat1243.html},
year = {2004},
date = {2004-01-01},
journal = {Nature Materials},
volume = {3},
number = {11},
pages = {779--782},
publisher = {Nature Publishing Group},
abstract = {A nanoscale understanding of the complex dynamics of large molecules at surfaces is essential for the bottom-up design of molecular nanostructures1, 2, 3, 4, 5, 6, 7, 8. Here we show that we can change the diffusion coefficient of the complex organic molecule known as Violet Lander (VL, C108H104) on Cu(110) by two orders of magnitude by using the STM at low temperatures to switch between two adsorption configurations that differ only in the molecular orientation with respect to the substrate lattice. From an interplay with molecular dynamics simulations, we interpret the results within a lock-and-key model similar to the one driving the recognition between biomolecules: the molecule (key) is immobilized only when its orientation is such that the molecular shape fits the atomic lattice of the surface (lock); otherwise the molecule is highly mobile.
Introduction
},
keywords = {Landers, Molecular Dynamics, Molecular Electronics, STM, top20},
pubstate = {published},
tppubtype = {article}
}
Introduction
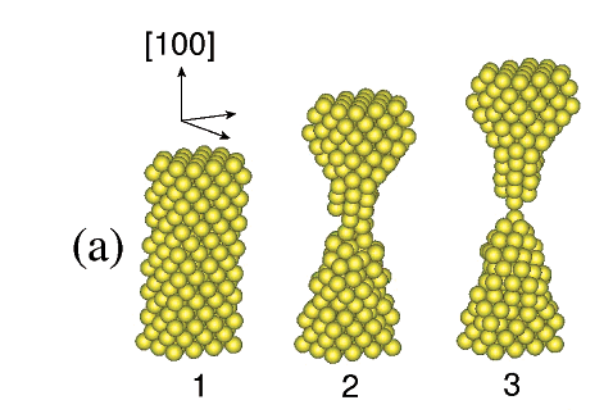
Coura, Pablo Z; Legoas, Sergio B; Moreira, Anderson S; Sato, Fernando; Rodrigues, Varlei; Dantas, Socrates O; Ugarte, Daniel; Galvao, Douglas S
On the structural and stability features of linear atomic suspended chains formed from gold nanowires stretching Journal Article
In: Nano Letters, vol. 4, no. 7, pp. 1187–1191, 2004.
Abstract | Links | BibTeX | Tags: Liinear Atomic Chains, Metallic Nanowires, Molecular Dynamics, Structure, TEM
@article{coura2004structural,
title = {On the structural and stability features of linear atomic suspended chains formed from gold nanowires stretching},
author = {Coura, Pablo Z and Legoas, Sergio B and Moreira, Anderson S and Sato, Fernando and Rodrigues, Varlei and Dantas, Socrates O and Ugarte, Daniel and Galvao, Douglas S},
url = {http://pubs.acs.org/doi/abs/10.1021/nl049725h},
year = {2004},
date = {2004-01-01},
journal = {Nano Letters},
volume = {4},
number = {7},
pages = {1187--1191},
publisher = {American Chemical Society},
abstract = {Metallic nanowires (NWs) have been intensily investigated in the past years, but details on
their formation are still not completely understood. In this work we report high resolution
transmission electron microscopy data and molecular dynamics simulation results for gold
NW elongation. Our results show that different initial crystallographic orientations lead to
very differentiated linear atomic suspended chain (LAC) formations and strongly support that
kinetic aspects are the dominant mechanisms determining the LAC morphologies.},
keywords = {Liinear Atomic Chains, Metallic Nanowires, Molecular Dynamics, Structure, TEM},
pubstate = {published},
tppubtype = {article}
}
their formation are still not completely understood. In this work we report high resolution
transmission electron microscopy data and molecular dynamics simulation results for gold
NW elongation. Our results show that different initial crystallographic orientations lead to
very differentiated linear atomic suspended chain (LAC) formations and strongly support that
kinetic aspects are the dominant mechanisms determining the LAC morphologies.
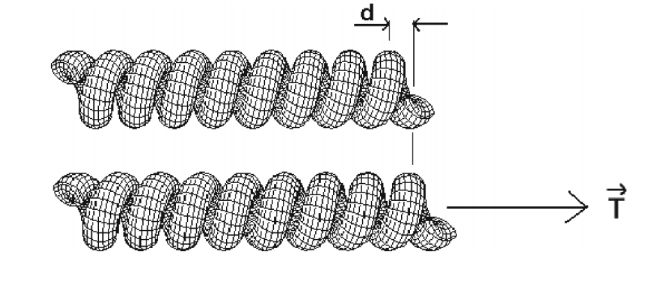
da Fonseca, Alexandre F; Galvao, Douglas S
Mechanical properties of nanosprings Journal Article
In: Physical review letters, vol. 92, no. 17, pp. 175502, 2004.
Abstract | Links | BibTeX | Tags: Elasticity, Filaments, Nanowires
@article{da2004mechanical,
title = {Mechanical properties of nanosprings},
author = {da Fonseca, Alexandre F and Galvao, Douglas S},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.92.175502},
year = {2004},
date = {2004-01-01},
journal = {Physical review letters},
volume = {92},
number = {17},
pages = {175502},
publisher = {APS},
abstract = {Nanostructures (nanotubes, nanowires, etc.) have been the object of intense theoretical and experimental investigations in recent years. Among these structures, helical nanosprings or nanocoils have attracted particular interest due to their special mechanical properties. In this work, we investigated structural properties of nanosprings in the Kirchhoff rod model. We derived expressions that can be used experimentally to obtain nanospring Young’s modulus and Poisson’s ratio values. Our results also might explain why the presence of catalytic particles is so important in nanostructure growth.},
keywords = {Elasticity, Filaments, Nanowires},
pubstate = {published},
tppubtype = {article}
}
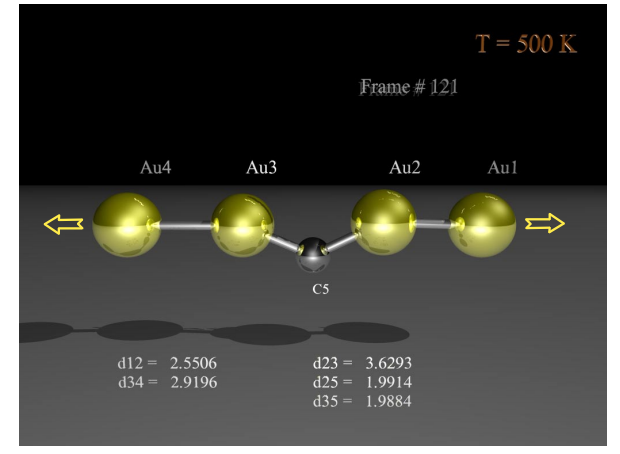
Legoas, Sergio B; Rodrigues, Varlei; Ugarte, Daniel; Galvao, Douglas S
Contaminants in suspended gold chains: An ab initio molecular dynamics study Journal Article
In: Physical Review Letters, vol. 93, no. 21, pp. 216103, 2004.
Abstract | Links | BibTeX | Tags: DFT, Electronic Structure, Linear Atomic Chains, Metallic Nanowires, top20
@article{legoas2004contaminants,
title = {Contaminants in suspended gold chains: An ab initio molecular dynamics study},
author = {Legoas, Sergio B and Rodrigues, Varlei and Ugarte, Daniel and Galvao, Douglas S},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.93.216103},
year = {2004},
date = {2004-01-01},
journal = {Physical Review Letters},
volume = {93},
number = {21},
pages = {216103},
publisher = {American Physical Society},
abstract = {Recently, we have proposed that the origin of anomalously long interatomic distances in suspended gold chains could be the result of carbon contamination during sample manipulation [S. B. Legoas et al., Phys. Rev. Lett. 88, 076105 (2002)]. More recently, however, other works have proposed that hydrogen instead of carbon should be the most probable contaminant. We report ab initio molecular dynamics results for different temperatures considering different possible contaminants. Our results show that at nonzero temperatures (more realistic to simulate the experimental conditions) hydrogen may be ruled out and carbon atoms remain the best candidate for contamination.},
keywords = {DFT, Electronic Structure, Linear Atomic Chains, Metallic Nanowires, top20},
pubstate = {published},
tppubtype = {article}
}
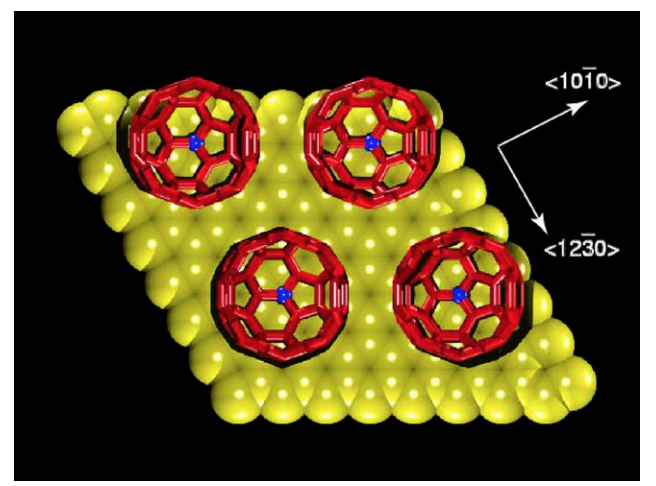
Legoas, Sergio B; Giro, Ronaldo; Galvao, Douglas S
Molecular dynamics simulations of C6) nanobearings Journal Article
In: Chemical physics letters, vol. 386, no. 4, pp. 425–429, 2004.
Abstract | Links | BibTeX | Tags: C60, Fullerenes, Molecular Dynamics, Nanobearing, Tribology
@article{legoas2004molecular,
title = {Molecular dynamics simulations of C6) nanobearings},
author = {Legoas, Sergio B and Giro, Ronaldo and Galvao, Douglas S},
url = {http://www.sciencedirect.com/science/article/pii/S000926140400168X},
year = {2004},
date = {2004-01-01},
journal = {Chemical physics letters},
volume = {386},
number = {4},
pages = {425--429},
publisher = {Elsevier},
abstract = {Recently was reported an ultra-lubricated system based on C60 molecules deposited over graphite layers. In that work a stick-slip rolling model for C60 molecules was proposed to explain the observed ultra-low friction force. In this Letter, we report the first molecular dynamics studies for these systems. Our results show that the AB stacking is not observed and the main experimental features can be explained without invoking stick-slip motions.
},
keywords = {C60, Fullerenes, Molecular Dynamics, Nanobearing, Tribology},
pubstate = {published},
tppubtype = {article}
}
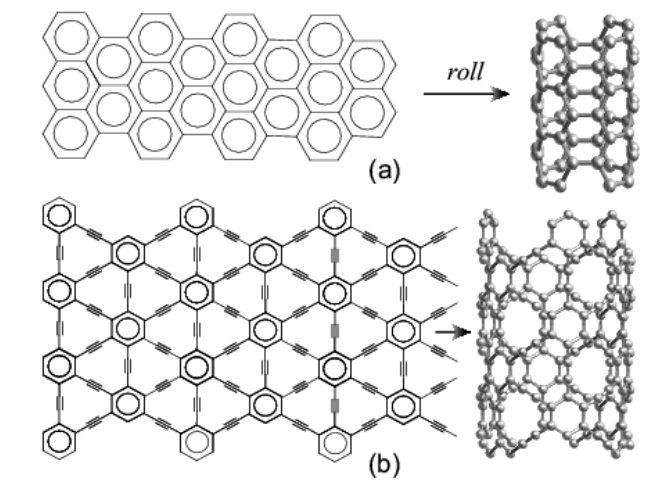
Coluci, VR; Braga, SF; Legoas, SB; Galvao, DS; Baughman, RH
New families of carbon nanotubes based on graphyne motifs Journal Article
In: Nanotechnology, vol. 15, no. 4, pp. S142, 2004.
Abstract | Links | BibTeX | Tags: Allotropes, Electronic Structure, Graphynes, Nanotubes
@article{coluci2004new,
title = {New families of carbon nanotubes based on graphyne motifs},
author = {Coluci, VR and Braga, SF and Legoas, SB and Galvao, DS and Baughman, RH},
url = {http://iopscience.iop.org/0957-4484/15/4/006},
year = {2004},
date = {2004-01-01},
journal = {Nanotechnology},
volume = {15},
number = {4},
pages = {S142},
publisher = {IOP Publishing},
abstract = {Electronic properties of proposed new families of carbon single walled nanotubes are investigated. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Analogous to ordinary nanotubes, armchair, zigzag and chiral graphyne nanotubes are possible. Tight-binding and ab initio density functional methods were used to predict the electronic properties of these unusual nanotubes. Of the three graphyne nanotube families analysed here, two provide metallic behaviour for armchair tubes and either metallic or semiconducting behaviour for zigzag nanotubes. For the other graphyne nanotube family investigated a diameter and chirality independent bandgap is predicted and a bandgap modulation study by structural distortions has been carried out for small longitudinal tube deformations. Interestingly, while the bandgap is insensitive to structure, the stress-induced bandgap changes can strongly depend both on the nanotube type and whether the strain is tensile or compressive.
},
keywords = {Allotropes, Electronic Structure, Graphynes, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
Campos, Paulo RA; de Oliveira, Viviane M; Giro, Ronaldo; Galvao, Douglas S
Emergence of prime numbers as the result of evolutionary strategy Journal Article
In: Physical Review Letters, vol. 93, no. 9, pp. 098107, 2004.
Abstract | Links | BibTeX | Tags: Cellular Automata, Cicadas, Patterns, Prime Numbers, top20
@article{campos2004emergence,
title = {Emergence of prime numbers as the result of evolutionary strategy},
author = {Campos, Paulo RA and de Oliveira, Viviane M and Giro, Ronaldo and Galvao, Douglas S},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.93.098107},
year = {2004},
date = {2004-01-01},
journal = {Physical Review Letters},
volume = {93},
number = {9},
pages = {098107},
publisher = {American Physical Society},
abstract = {We investigate by means of a simple theoretical model the emergence of prime numbers as life cycles, as those seen for some species of cicadas. The cicadas, more precisely the Magicicadas, spend most of their lives below the ground and then emerge and die in a short period of time. The Magicicadas display an uncommon behavior: their emergence is synchronized and these periods are usually prime numbers. In the current work, we develop a spatially extended model at which preys and predators coexist and can change their evolutionary dynamics through the occurrence of mutations. We verified that prime numbers as life cycles emerge as a result of the evolution of the population. Our results seem to be a first step in order to prove that the development of such strategy is selectively advantageous, especially for those organisms that are highly vulnerable to attacks of predators.},
keywords = {Cellular Automata, Cicadas, Patterns, Prime Numbers, top20},
pubstate = {published},
tppubtype = {article}
}
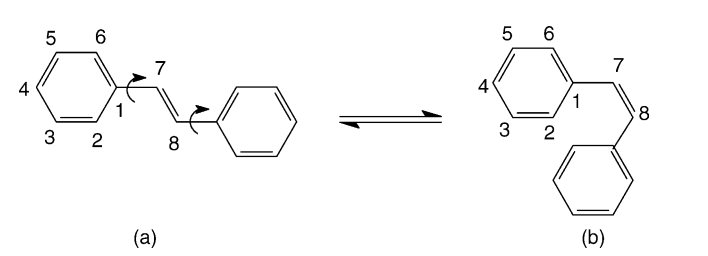
Vendrame, R; Coluci, Vitor Rafael; Galvao, Douglas Soares
Comparative parametric method 5 (PM5) study of trans-stilbene Journal Article
In: Journal of Molecular Structure: THEOCHEM, vol. 686, no. 1, pp. 103–108, 2004.
Abstract | Links | BibTeX | Tags: Electronic Structure, MOPAC, PM3, PM5, Stilbene
@article{vendrame2004comparative,
title = {Comparative parametric method 5 (PM5) study of trans-stilbene},
author = {Vendrame, R and Coluci, Vitor Rafael and Galvao, Douglas Soares},
url = {http://www.sciencedirect.com/science/article/pii/S0166128004005998},
year = {2004},
date = {2004-01-01},
journal = {Journal of Molecular Structure: THEOCHEM},
volume = {686},
number = {1},
pages = {103--108},
publisher = {Elsevier},
abstract = {In this work we report a comparative Austin method 1 (AM1), parametric method 3 (PM3), and parametric method 5 (PM5) studies for trans-stilbene in its ground, excited (singlet and triplet), and ionic (positive and negative polarons and bipolarons) states. We evaluated the accuracy of the recently developed PM5 method. PM5 and AM1 predict a non-planar ground and singlet states for trans-stilbene, while PM3 predicts planar ones, which is in agreement with the available experimental data. In general the PM3 and PM5 bond lengths are superior to AM1 while AM1 bond angles are superior to PM3 and PM5 when compared with available experimental data. The PM5 underestimates the cis–trans isomerization energy and and it is not a quite reliable method for the calculation of relative IP values. The presumed PM5 superior performance against AM1 and PM3 was not observed for the stilbene structures.
},
keywords = {Electronic Structure, MOPAC, PM3, PM5, Stilbene},
pubstate = {published},
tppubtype = {article}
}

Coluci, VR; Galvao, DS; Baughman, RH
Theoretical investigation of electromechanical effects for graphyne carbon nanotubes Journal Article
In: The Journal of chemical physics, vol. 121, no. 7, pp. 3228–3237, 2004.
Abstract | Links | BibTeX | Tags: Allotropes, Electroactuation, Electronic Structure, Graphynes, Nanotubes
@article{coluci2004theoretical,
title = {Theoretical investigation of electromechanical effects for graphyne carbon nanotubes},
author = {Coluci, VR and Galvao, DS and Baughman, RH},
url = {http://scitation.aip.org/content/aip/journal/jcp/121/7/10.1063/1.1772756},
year = {2004},
date = {2004-01-01},
journal = {The Journal of chemical physics},
volume = {121},
number = {7},
pages = {3228--3237},
publisher = {AIP Publishing},
abstract = {We present a theoretical study of the electronic and mechanical properties of graphyne-based nanotubes (GNTs). These semiconducting nanotubes result from the elongation of one-third of the covalent interconnections of graphite-based nanotubes by the introduction of yne groups. The effect of charge injection on the dimensions of GNTs was investigated using tight-binding calculations. Low amounts of electron injection are predicted to cause qualitatively different responses for armchair and zigzag graphyne nanotubes. Although the behavior is qualitatively similar to the usual carbon nanotubes, the charge-induced strains are predicted to be smaller for the GNTs than for ordinary single walled carbon nanotubes.},
keywords = {Allotropes, Electroactuation, Electronic Structure, Graphynes, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
![Benzo [c] quinolizin-3-ones theoretical investigation: SAR analysis and application to nontested compounds](https://sites.ifi.unicamp.br/galvao/files/2015/02/Screen-Shot-2015-02-26-at-12.21.40-PM.png)
Braga, Scheila Furtado; Galvao, Douglas Soares
Benzo [c] quinolizin-3-ones theoretical investigation: SAR analysis and application to nontested compounds Journal Article
In: Journal of chemical information and computer sciences, vol. 44, no. 6, pp. 1987–1997, 2004.
Abstract | Links | BibTeX | Tags: Drug Design, Electronic Structure, PCA/HCA, Theory of Electronic Indices
@article{braga2004benzo,
title = {Benzo [c] quinolizin-3-ones theoretical investigation: SAR analysis and application to nontested compounds},
author = {Braga, Scheila Furtado and Galvao, Douglas Soares},
url = {http://pubs.acs.org/doi/abs/10.1021/ci049837u},
year = {2004},
date = {2004-01-01},
journal = {Journal of chemical information and computer sciences},
volume = {44},
number = {6},
pages = {1987--1997},
publisher = {American Chemical Society},
abstract = {We investigate with the use of theoretical methodologies the activity of a set of 41 benzo[c]quinolizin-3-
ones (BC3), some of them explored as selective inhibitors of the human 5R-reductase steroid. For the
structure-activity study we have considered dividing the molecules into groups of tested and nontested
compounds. Semiempirical calculations and pattern recognition methods such as Electronic Indices
Methodology (EIM), Principal Components Analysis (PCA), Hierarchical Cluster Analysis (HCA), and
K-Nearest Neighbors (KNN) have been applied to search for a correlation between experimental activity
and theoretical descriptors. Our results show that it is possible to directly correlate some molecular quantum
descriptors with BC3 biological activity. This information can be used in principle to identify active/inactive
untested compounds and/or to design new active compounds.},
keywords = {Drug Design, Electronic Structure, PCA/HCA, Theory of Electronic Indices},
pubstate = {published},
tppubtype = {article}
}
ones (BC3), some of them explored as selective inhibitors of the human 5R-reductase steroid. For the
structure-activity study we have considered dividing the molecules into groups of tested and nontested
compounds. Semiempirical calculations and pattern recognition methods such as Electronic Indices
Methodology (EIM), Principal Components Analysis (PCA), Hierarchical Cluster Analysis (HCA), and
K-Nearest Neighbors (KNN) have been applied to search for a correlation between experimental activity
and theoretical descriptors. Our results show that it is possible to directly correlate some molecular quantum
descriptors with BC3 biological activity. This information can be used in principle to identify active/inactive
untested compounds and/or to design new active compounds.
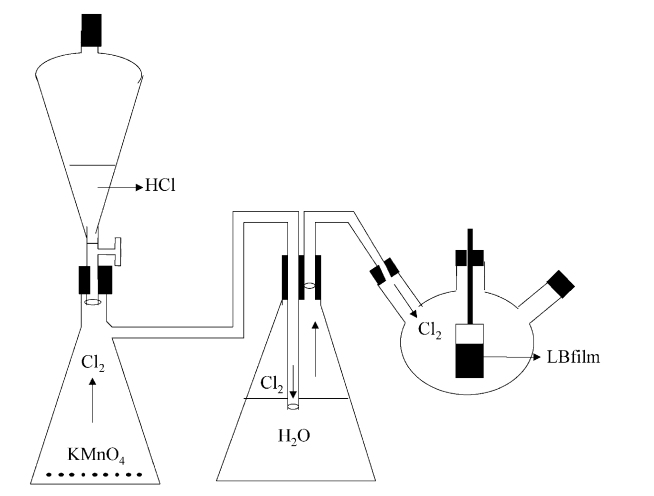
Gaffo, Luciana; Couto Jr, Odilon DD; Giro, Ronaldo; Brasil, Maria JSP; Galvao, Douglas S; Cerdeira, Fernando; de Oliveira Jr, Osvaldo N; Wohnrath, Karen
Effects of chlorine gas exposure on the optical properties of rhodium phthalocyanine films Journal Article
In: Solid state communications, vol. 131, no. 1, pp. 53–56, 2004.
Abstract | Links | BibTeX | Tags: Films, Phthalocyanine, Sensors
@article{gaffo2004effects,
title = {Effects of chlorine gas exposure on the optical properties of rhodium phthalocyanine films},
author = {Gaffo, Luciana and Couto Jr, Odilon DD and Giro, Ronaldo and Brasil, Maria JSP and Galvao, Douglas S and Cerdeira, Fernando and de Oliveira Jr, Osvaldo N and Wohnrath, Karen},
url = {http://www.sciencedirect.com/science/article/pii/S0038109804002996},
year = {2004},
date = {2004-01-01},
journal = {Solid state communications},
volume = {131},
number = {1},
pages = {53--56},
publisher = {Pergamon},
abstract = {We investigated the effects of exposing rhodium phthalocyanine films deposited on glass substrates by the Lagmuir–Blodgett technique to chlorine gas. The visual aspect of the films is altered upon chlorination, changing in color from blue to transparent. We performed optical absorption and Raman Scattering measurements on our films prior to and after exposing it to chlorine gas. We observed a pronounced quenching of the characteristic triplet centered around the Q-absorption band at 662 nm as a result of chlorine incorporation. Another absorption band, in the near UV part of the spectrum, is not greatly affected by the process. No new optical structures appear as a consequence of chlorination. Equivalent effects were observed in the Raman spectra. Leaving the previously exposed films in air for several hours results in a slow partial recovery of the optical spectra. This recovery, as well as the amount of original quenching, depends on the amount of time during which the film was exposed to chlorine.},
keywords = {Films, Phthalocyanine, Sensors},
pubstate = {published},
tppubtype = {article}
}
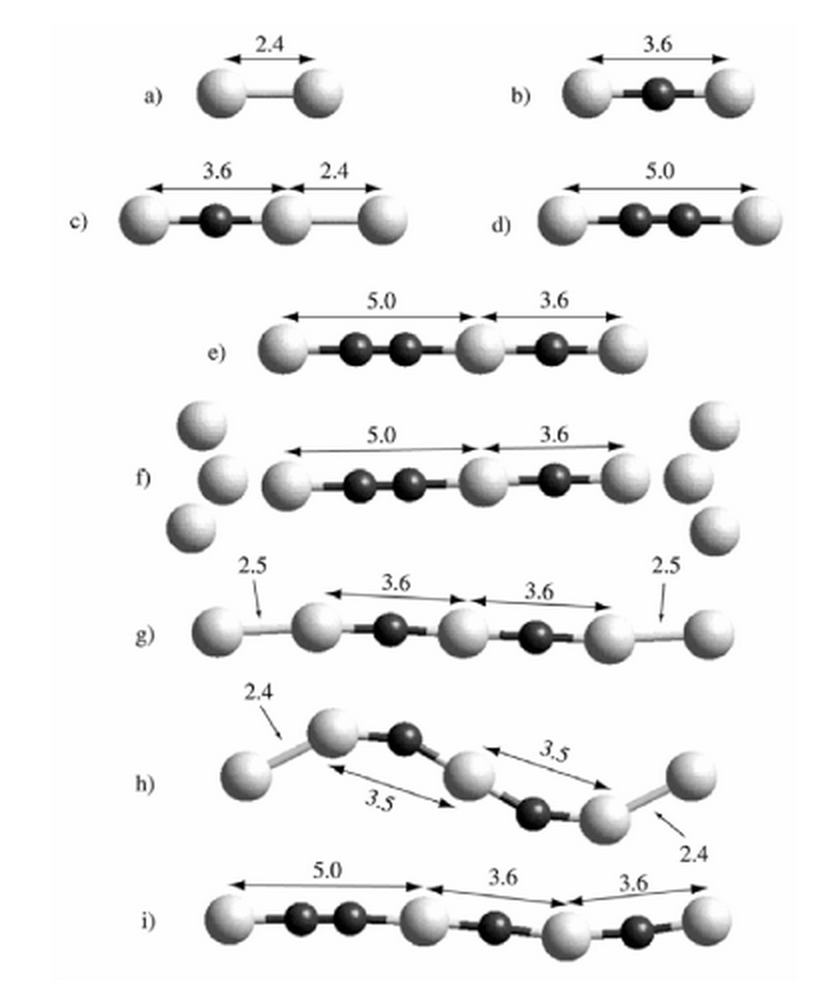
Galvao, Douglas Soares; Rodrigues, Varlei; Ugarte, Daniel; Legoas, Sergio Benites
The role of carbon contamination in metallic nanowires Journal Article
In: Materials Research, vol. 7, no. 2, pp. 339–342, 2004.
Abstract | Links | BibTeX | Tags: Contaminantes, Electronic Structure, Linear Atomic Chains, Metallic Nanowires, Structure
@article{galvao2004role,
title = {The role of carbon contamination in metallic nanowires},
author = {Galvao, Douglas Soares and Rodrigues, Varlei and Ugarte, Daniel and Legoas, Sergio Benites},
url = {http://www.scielo.br/scielo.php?pid=S1516-14392004000200020&script=sci_arttext},
year = {2004},
date = {2004-01-01},
journal = {Materials Research},
volume = {7},
number = {2},
pages = {339--342},
publisher = {SciELO Brasil},
abstract = {Metallic nanowires have attracted much attention in the last years due to new phenomena such as quantum conductance and the existence of unexpected long interatomic distances attaining 0.3-0.5 nm. These large distances represented a challenge for physical interpretation. In this work we present experimental data from high-resolution transmission electron microscopy and results from ab initio calculations for suspended gold chains and show that these large distances can be easily explained by the presence of carbon atoms as contaminants. In principle the present conclusions can be also applied to other metallic nanowires (such as Ag and Pt) whose structures also present large interatomic distances.},
keywords = {Contaminantes, Electronic Structure, Linear Atomic Chains, Metallic Nanowires, Structure},
pubstate = {published},
tppubtype = {article}
}
dos Santos, Adenilson O; Avanci, Luis H; Cardoso, Lisandro P; Giro, Ronaldo; Legoas, Sergio B; Galvao, Douglas S; Sherwood, John N
Hysteresis-like behavior in MBANP crystals Journal Article
In: Crystal growth & design, vol. 4, no. 5, pp. 1079–1081, 2004.
Abstract | Links | BibTeX | Tags: Crystallographic Structure, Electronic Structure, Molecular Crystals
@article{dos2004hysteresis,
title = {Hysteresis-like behavior in MBANP crystals},
author = {dos Santos, Adenilson O and Avanci, Luis H and Cardoso, Lisandro P and Giro, Ronaldo and Legoas, Sergio B and Galvao, Douglas S and Sherwood, John N},
url = {http://pubs.acs.org/doi/abs/10.1021/cg0341860},
year = {2004},
date = {2004-01-01},
journal = {Crystal growth & design},
volume = {4},
number = {5},
pages = {1079--1081},
publisher = {ACS Publications},
abstract = {he measured strain versus E-cycle in a MBANP organic single crystal showed interesting butterfly wing shape hysteresis behavior. Quantum mechanical calculations on isolated MBANP molecules have shown that the main features of the hysteresis shape can be explained in terms of field induced changes in the charge profiles and geometry of isolated MBANP molecules.},
keywords = {Crystallographic Structure, Electronic Structure, Molecular Crystals},
pubstate = {published},
tppubtype = {article}
}
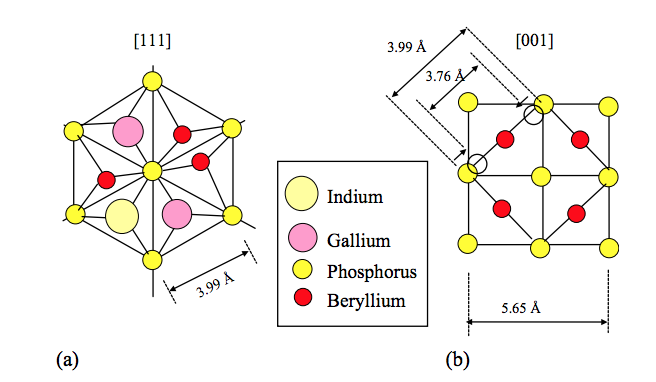
De Castro, MPP; Von Zuben, AA; Frateschi, NC; Santo, LLE; Galvao, DS; Bettini, J; De Carvalho, MMG
Strong spatial beryllium doping selectivity on InGaP layers grown on pre-patterned GaAs substrates by chemical beam epitaxy Journal Article
In: Journal of crystal growth, vol. 266, no. 4, pp. 429–434, 2004.
Abstract | Links | BibTeX | Tags: Crystal Growth, Doping, Electronic Structure
@article{de2004strong,
title = {Strong spatial beryllium doping selectivity on InGaP layers grown on pre-patterned GaAs substrates by chemical beam epitaxy},
author = {De Castro, MPP and Von Zuben, AA and Frateschi, NC and Santo, LLE and Galvao, DS and Bettini, J and De Carvalho, MMG},
url = {http://www.sciencedirect.com/science/article/pii/S0022024804002829},
year = {2004},
date = {2004-01-01},
journal = {Journal of crystal growth},
volume = {266},
number = {4},
pages = {429--434},
publisher = {North-Holland},
abstract = {We present an investigation of beryllium doping selectivity in InGaP layers grown by chemical beam epitaxy on pre-patterned substrates. We observed a resistivity of 3.1×10−2 and 4.5×10−2 Ω cm for (1 1 1)A planes with the growth at 500°C and 540°C, respectively. The layers on (0 0 1) planes show a resistivity of 8.9×10−1 Ω cm with the growth at 500°C, being essentially undoped with the growth at 540°C ⋅ We show how this strong doping selectivity can be explained by Be3P2 cluster formation growth, which depends on growth temperature and planar crystalline structure.},
keywords = {Crystal Growth, Doping, Electronic Structure},
pubstate = {published},
tppubtype = {article}
}
2003
Coluci, VR; Braga, SF; Legoas, SB; Galvao, DS; Baughman, RH
Families of carbon nanotubes: Graphyne-based nanotubes Journal Article
In: Physical Review B, vol. 68, no. 3, pp. 035430, 2003.
Abstract | Links | BibTeX | Tags: Allotropes, Electronic Structure, Graphynes, Nanotubes
@article{coluci2003families,
title = {Families of carbon nanotubes: Graphyne-based nanotubes},
author = {Coluci, VR and Braga, SF and Legoas, SB and Galvao, DS and Baughman, RH},
url = {http://journals.aps.org/prb/abstract/10.1103/PhysRevB.68.035430},
year = {2003},
date = {2003-01-01},
journal = {Physical Review B},
volume = {68},
number = {3},
pages = {035430},
publisher = {APS},
abstract = {New families of carbon single-walled nanotubes are proposed and their electronic structures are investigated. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Analogously to ordinary nanotubes, armchair, zigzag, and chiral graphyne nanotubes are possible. We here predict the electronic properties of these unusual nanotubes using tight-binding and ab initio density functional methods. Of the three graphyne nanotube families analyzed here, two provide metallic behavior for armchair tubes and either metallic or semiconducting behavior for zigzag nanotubes. A diameter- and chirality-independent band gap is predicted for the other investigated graphyne family, as well as an oscillatory dependence of the effective mass on nanotube diameter.},
keywords = {Allotropes, Electronic Structure, Graphynes, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
Braga, Scheila Furtado; Galvao, Douglas Soares
A structure-activity study of taxol, taxotere, and derivatives using the electronic indices methodology (EIM) Journal Article
In: Journal of chemical information and computer sciences, vol. 43, no. 2, pp. 699–706, 2003.
Abstract | Links | BibTeX | Tags: Drug Design, Electronic Structure, Taxol, Taxotere, Theory of Electronic Indices
@article{braga2003structure,
title = {A structure-activity study of taxol, taxotere, and derivatives using the electronic indices methodology (EIM)},
author = {Braga, Scheila Furtado and Galvao, Douglas Soares},
url = {http://pubs.acs.org/doi/abs/10.1021/ci025640v},
year = {2003},
date = {2003-01-01},
journal = {Journal of chemical information and computer sciences},
volume = {43},
number = {2},
pages = {699--706},
publisher = {American Chemical Society},
abstract = {Among the new families of effective anticancer drugs, the natural product paclitaxel (Taxol/Bristol-MyersSquibb)
and its semisynthetic derivative docetaxel (Taxotere/Rhone-Poulenc Rorer) are probably the most
promising agents under investigation. Surprisingly considering their importance no detailed quantum
mechanical studies have been carried out for these drugs. In this work we report the first structure-activity
relationship (SAR) studies for 20 taxoid structures using molecular descriptors from all-electron quantum
methods. The used methods were the pattern-recognition Principal Component Analysis (PCA), Hierarchical
Clustering Analysis (HCA), and the recently developed Electronic Indices Methodology (EIM). The combined
use of EIM with PCA/HCA methodologies was able to correctly classify active and inactive taxoids with
100% of accuracy using only a few “universal” quantum molecular descriptors. It was possible to identify
the electronic features defining active molecules. This information can be used to select and design new
active compounds. The combined use of EIM with PCA/HCA can be a new and very efficient tool in the
field of computer assisted drug design.},
keywords = {Drug Design, Electronic Structure, Taxol, Taxotere, Theory of Electronic Indices},
pubstate = {published},
tppubtype = {article}
}
and its semisynthetic derivative docetaxel (Taxotere/Rhone-Poulenc Rorer) are probably the most
promising agents under investigation. Surprisingly considering their importance no detailed quantum
mechanical studies have been carried out for these drugs. In this work we report the first structure-activity
relationship (SAR) studies for 20 taxoid structures using molecular descriptors from all-electron quantum
methods. The used methods were the pattern-recognition Principal Component Analysis (PCA), Hierarchical
Clustering Analysis (HCA), and the recently developed Electronic Indices Methodology (EIM). The combined
use of EIM with PCA/HCA methodologies was able to correctly classify active and inactive taxoids with
100% of accuracy using only a few “universal” quantum molecular descriptors. It was possible to identify
the electronic features defining active molecules. This information can be used to select and design new
active compounds. The combined use of EIM with PCA/HCA can be a new and very efficient tool in the
field of computer assisted drug design.
Giro, Ronaldo; Cyrillo, Marcio; Galvao, Douglas Soares
Using artificial intelligence methods to design new conducting polymers Journal Article
In: Materials Research, vol. 6, no. 4, pp. 523–528, 2003.
Abstract | Links | BibTeX | Tags: Artificial Intelligence, Conducting Polymers, Genetic Algorithms, Material Design
@article{giro2003using,
title = {Using artificial intelligence methods to design new conducting polymers},
author = {Giro, Ronaldo and Cyrillo, Marcio and Galvao, Douglas Soares},
url = {http://www.scielo.br/scielo.php?pid=S1516-14392003000400017&script=sci_arttext},
year = {2003},
date = {2003-01-01},
journal = {Materials Research},
volume = {6},
number = {4},
pages = {523--528},
publisher = {SciELO Brasil},
abstract = {In the last years the possibility of creating new conducting polymers exploring the concept of copolymerization (different structural monomeric units) has attracted much attention from experimental and theoretical points of view. Due to the rich carbon reactivity an almost infinite number of new structures is possible and the procedure of trial and error has been the rule. In this work we have used a methodology able of generating new structures with pre-specified properties. It combines the use of negative factor counting (NFC) technique with artificial intelligence methods (genetic algorithms - GAs). We present the results for a case study for poly(phenylenesulfide phenyleneamine) (PPSA), a copolymer formed by combination of homopolymers: polyaniline (PANI) and polyphenylenesulfide (PPS). The methodology was successfully applied to the problem of obtaining binary up to quinternary disordered polymeric alloys with a pre-specific gap value or exhibiting metallic properties. It is completely general and can be in principle adapted to the design of new classes of materials with pre-specified properties},
keywords = {Artificial Intelligence, Conducting Polymers, Genetic Algorithms, Material Design},
pubstate = {published},
tppubtype = {article}
}
Del Nero, J; Galvao, DS; Laks, B
Electronic structure investigation of biosensor polymer Journal Article
In: Optical Materials, vol. 21, no. 1, pp. 461–466, 2003.
Abstract | Links | BibTeX | Tags: Conducting Polymers, Electronic Structure, Sensors
@article{del2003electronic,
title = {Electronic structure investigation of biosensor polymer},
author = {Del Nero, J and Galvao, DS and Laks, B},
url = {http://www.sciencedirect.com/science/article/pii/S0925346702001830},
year = {2003},
date = {2003-01-01},
journal = {Optical Materials},
volume = {21},
number = {1},
pages = {461--466},
publisher = {Elsevier},
abstract = {We report a theoretical study of the ground, excited and ionic states of 3-methyl pyrrole-4-carboxilic acid (MPC) oligomers and related compounds which present conformational defects. Our results reveal the existence of differentiated electronic behavior for MPC with relation to oligopyrrole derivatives. These electronic features might explain why MPC works properly as a biosensor for cytochrome C while no voltametric response is observed for unsubstituted poly(pyrrole).},
keywords = {Conducting Polymers, Electronic Structure, Sensors},
pubstate = {published},
tppubtype = {article}
}
Giro, R; Galvao, DS
Semiempirical studies of the electronic structure of polyphenylene sulfide phenyleneamine Journal Article
In: International journal of quantum chemistry, vol. 95, no. 3, pp. 252–259, 2003.
Abstract | Links | BibTeX | Tags: co-polymers, Conducting Polymers, Electronic Structure, PANi, PPS, PPSA
@article{giro2003semiempirical,
title = {Semiempirical studies of the electronic structure of polyphenylene sulfide phenyleneamine},
author = {Giro, R and Galvao, DS},
url = {http://onlinelibrary.wiley.com/doi/10.1002/qua.10722/full},
year = {2003},
date = {2003-01-01},
journal = {International journal of quantum chemistry},
volume = {95},
number = {3},
pages = {252--259},
publisher = {Wiley Subscription Services, Inc., A Wiley Company},
abstract = {Polyphenylene sulfide (PPS) and polyaniline (PANI) are heteroatom-containing polymers with unique properties. While PPS provides a good level of chemical and thermal stability, PANI is important due to its ability to form electrically conducting films. A combination of PPS and PANI, with alternating phenyleneamine and phenylene sulfide blocks, resulted in a new material combining the structural features of PPS and PANI. This copolymer is known as polyphenylene sulfide–phenyleneamine (PPSA). In this work we present geometric and spectroscopic studies on PPSA oligomers using the well-known semiempirical methods PM3 (parametric method 3) and ZINDO-S/CI (Zerner's intermediate neglect of differential overlap/spectroscopic-configuration interaction). PM3 results show that long PPSA oligomers present alternating in- and out-of-plane rings with torsion angles about 60°. The ZINDO-simulated spectra compare well with the available experimental data. The origin of the low PPSA conductivity is addressed in terms of electronic features presented by isolated polymeric chains. © 2003 Wiley Periodicals, Inc. Int J Quantum Chem 95: 252–259, 2003},
keywords = {co-polymers, Conducting Polymers, Electronic Structure, PANi, PPS, PPSA},
pubstate = {published},
tppubtype = {article}
}
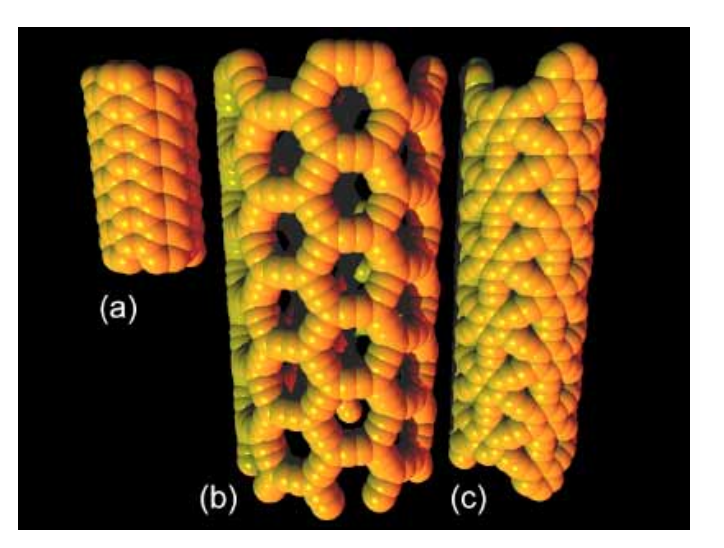
Coluci, VR; Braga, SF; Legoas, SB; Galvao, DS; Baughman, RH
Graphyne Nanotubes: New Families of Carbon Nanotubes Proceedings
Warrendale, Pa.; Materials Research Society; 1999, vol. 739, 2003.
Abstract | Links | BibTeX | Tags: Allotropes, Electronic Structure, Graphynes, Nanotubes
@proceedings{coluci2003graphyne,
title = {Graphyne Nanotubes: New Families of Carbon Nanotubes},
author = {Coluci, VR and Braga, SF and Legoas, SB and Galvao, DS and Baughman, RH},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8031794},
year = {2003},
date = {2003-01-01},
journal = {MATERIALS RESEARCH SOCIETY SYMPOSIUM PROCEEDINGS},
volume = {739},
pages = {175--180},
publisher = {Warrendale, Pa.; Materials Research Society; 1999},
abstract = {Fundamentally new families of carbon single walled nanotubes are proposed. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Similarly to ordinary nanotubes, armchair, zig-zag, and chiral graphyne nanotubes are possible. We present here results for the electronic properties of graphyne based tubes obtained from tight-binding and ab initio density functional methods.},
keywords = {Allotropes, Electronic Structure, Graphynes, Nanotubes},
pubstate = {published},
tppubtype = {proceedings}
}

VR Coluci SB Legoas, SF Braga
Molecular-dynamics simulations of carbon nanotubes as gigahertz oscillators Journal Article
In: Physical Review Letters, vol. 90, no. 5, pp. 055504, 2003.
Abstract | BibTeX | Tags: Carbon Nanotubes, Molecular Dynamics, Oscillators, top20
@article{legoas2003molecularb,
title = {Molecular-dynamics simulations of carbon nanotubes as gigahertz oscillators},
author = {SB Legoas, VR Coluci, SF Braga, PZ Coura, SO Dantas, DS Galvao},
year = {2003},
date = {2003-01-01},
journal = {Physical Review Letters},
volume = {90},
number = {5},
pages = {055504},
abstract = {Recently, Zheng and Jiang [Phys. Rev. Lett. 88, 045503 (2002)] have proposed that multiwalled carbon nanotubes could be the basis for a new generation of nano-oscillators in the several gigahertz range. In this Letter, we present the first molecular dynamics simulation for these systems. Different nanotube types were considered in order to verify the reliability of such devices as gigahertz oscillators. Our results show that these nano-oscillators are dynamically stable when the radii difference values between inner and outer tubes are of ∼3.4 Å. Frequencies as large as 38 GHz were observed, and the calculated force values are in good agreement with recent experimental investigations.},
keywords = {Carbon Nanotubes, Molecular Dynamics, Oscillators, top20},
pubstate = {published},
tppubtype = {article}
}
2002

Legoas, Sergio B; Galvao, Douglas S; Rodrigues, Varlei; Ugarte, Daniel
Origin of anomalously long interatomic distances in suspended gold chains Journal Article
In: Physical Review Letters, vol. 88, no. 7, pp. 076105, 2002.
Abstract | Links | BibTeX | Tags: DFT, Electronic Structure, Linear Atomic Chains, Metallic Nanowires, TEM, top20
@article{legoas2002origin,
title = {Origin of anomalously long interatomic distances in suspended gold chains},
author = {Legoas, Sergio B and Galvao, Douglas S and Rodrigues, Varlei and Ugarte, Daniel},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.88.076105},
year = {2002},
date = {2002-01-01},
journal = {Physical Review Letters},
volume = {88},
number = {7},
pages = {076105},
publisher = {American Physical Society},
abstract = {The discovery of long bonds in gold atom chains has represented a challenge for physical interpretation. In fact, interatomic distances frequently attain 3.0–3.6 Å values, and distances as large as 5.0 Å may be occasionally observed. Here we studied gold chains by transmission electron microscopy and performed theoretical calculations using cluster ab initio density functional formalism. We show that the insertion of two carbon atoms is required to account for the longest bonds, while distances above 3 Å may be due to a mixture of clean and one C atom contaminated bonds.},
keywords = {DFT, Electronic Structure, Linear Atomic Chains, Metallic Nanowires, TEM, top20},
pubstate = {published},
tppubtype = {article}
}
Giro, R; Cyrillo, M; Galvao, DS
Designing conducting polymers using genetic algorithms Journal Article
In: Chemical Physics Letters, vol. 366, no. 1, pp. 170–175, 2002.
Abstract | Links | BibTeX | Tags: Conducting Polymers, Electronic Structure, Genetic Algorithms, Material Design
@article{giro2002designing,
title = {Designing conducting polymers using genetic algorithms},
author = {Giro, R and Cyrillo, M and Galvao, DS},
url = {http://www.sciencedirect.com/science/article/pii/S0009261402015476},
year = {2002},
date = {2002-01-01},
journal = {Chemical Physics Letters},
volume = {366},
number = {1},
pages = {170--175},
publisher = {North-Holland},
abstract = {We have developed a new methodology to design conducting polymers with pre-specified properties. The methodology is based on the use of genetic algorithms (GAs) coupled to Negative Factor Counting technique. We present the results for a case study of polyanilines, one of the most important families of conducting polymers. The methodology proved to be able of generating automatic solutions for the problem of determining the optimum relative concentration for binary and ternary disordered polyaniline alloys exhibiting metallic properties. The methodology is completely general and can be used to design new classes of materials.},
keywords = {Conducting Polymers, Electronic Structure, Genetic Algorithms, Material Design},
pubstate = {published},
tppubtype = {article}
}
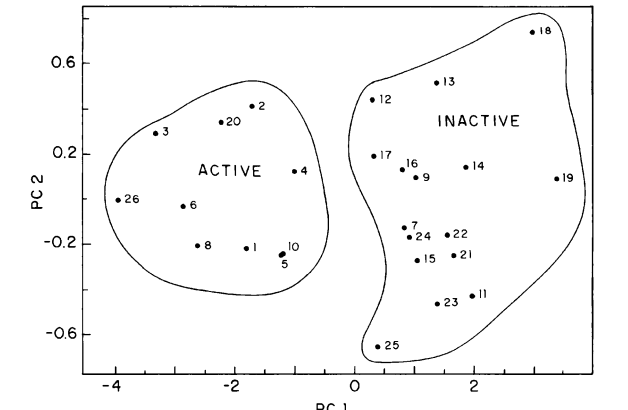
Coluci, Vitor Rafael; Vendrame, Rosana; Braga, RS; Galvao, DS
In: Journal of chemical information and computer sciences, vol. 42, no. 6, pp. 1479–1489, 2002.
Abstract | Links | BibTeX | Tags: Carcinogenesis, Neural Networks, PCA/HCA, Polycyclic Aromatic Hydrocarbons (PAHs), Theory of Electronic Indices
@article{coluci2002identifying,
title = {Identifying relevant molecular descriptors related to carcinogenic activity of polycyclic aromatic hydrocarbons (PAHs) using pattern recognition methods},
author = {Coluci, Vitor Rafael and Vendrame, Rosana and Braga, RS and Galvao, DS},
url = {http://pubs.acs.org/doi/abs/10.1021/ci025577%2B},
year = {2002},
date = {2002-01-01},
journal = {Journal of chemical information and computer sciences},
volume = {42},
number = {6},
pages = {1479--1489},
publisher = {American Chemical Society},
abstract = {Polycyclic Aromatic Hydrocarbons (PAHs) constitute an important family of molecules capable of inducing
chemical carcinogenesis. In this work we report structure-activity relationship (SAR) studies for 81 PAHs
using the pattern-recognition methods Principal Component Analysis (PCA), Hierarchical Clustering Analysis
(HCA) and Neural Networks (NN). The used molecular descriptors were obtained from the semiempirical
Parametric Method 3 (PM3) calculations. We have developed a new procedure that is capable of identifying
the PAHs’ carcinogenic activity with an accuracy higher than 80%. PCA selected molecular descriptors
that can be directly correlated with some models proposed to PAHs’ metabolic activation mechanism leading
to the formation of PAHs-DNA adducts. PCA, HCA and NN validate the energy separation between the
highest occupied molecular orbital and its next lower level as a major descriptor defining the carcinogenic
activity. This descriptor has been only recently discussed in the literature as one new possible universal
parameter for defining the biological activity of several classes of compounds.},
keywords = {Carcinogenesis, Neural Networks, PCA/HCA, Polycyclic Aromatic Hydrocarbons (PAHs), Theory of Electronic Indices},
pubstate = {published},
tppubtype = {article}
}
chemical carcinogenesis. In this work we report structure-activity relationship (SAR) studies for 81 PAHs
using the pattern-recognition methods Principal Component Analysis (PCA), Hierarchical Clustering Analysis
(HCA) and Neural Networks (NN). The used molecular descriptors were obtained from the semiempirical
Parametric Method 3 (PM3) calculations. We have developed a new procedure that is capable of identifying
the PAHs’ carcinogenic activity with an accuracy higher than 80%. PCA selected molecular descriptors
that can be directly correlated with some models proposed to PAHs’ metabolic activation mechanism leading
to the formation of PAHs-DNA adducts. PCA, HCA and NN validate the energy separation between the
highest occupied molecular orbital and its next lower level as a major descriptor defining the carcinogenic
activity. This descriptor has been only recently discussed in the literature as one new possible universal
parameter for defining the biological activity of several classes of compounds.
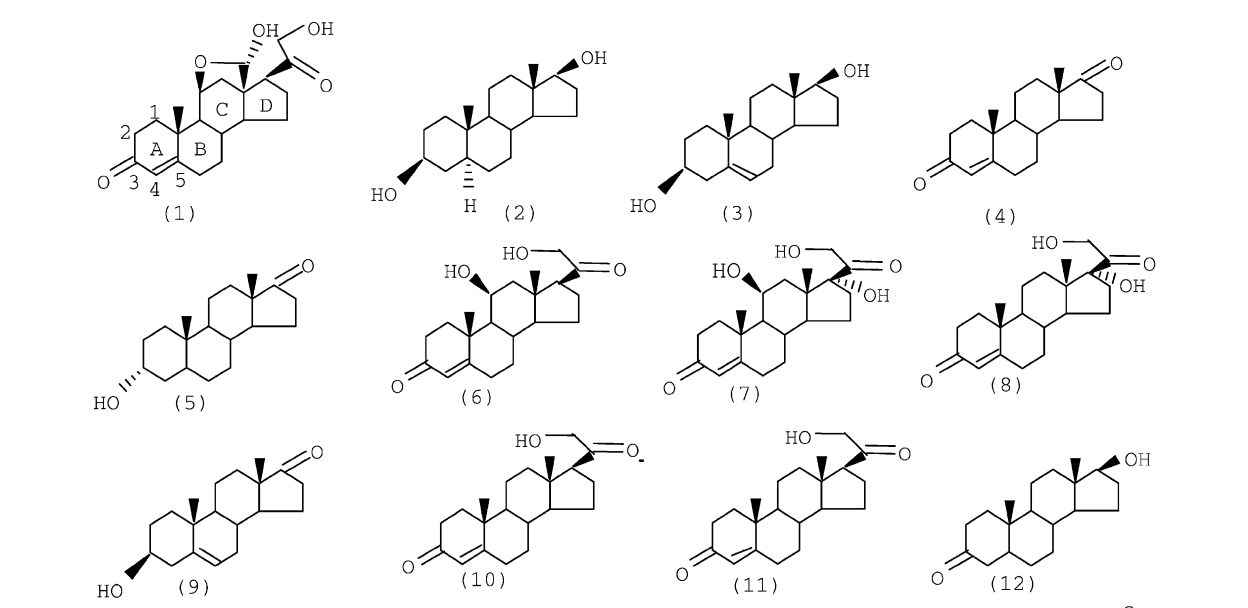
Vendrame, R; Coluci, VR; Braga, RS; Galvao, DS
Structure--activity relationship (SAR) studies of the tripos benchmark steroids Journal Article
In: Journal of Molecular Structure: THEOCHEM, vol. 619, no. 1, pp. 195–205, 2002.
Abstract | Links | BibTeX | Tags: Drug Design, Electronic Structure, HCA/PCA, Neural Networks, Tripos
@article{vendrame2002structure,
title = {Structure--activity relationship (SAR) studies of the tripos benchmark steroids},
author = {Vendrame, R and Coluci, VR and Braga, RS and Galvao, DS},
url = {http://www.sciencedirect.com/science/article/pii/S016612800200578X},
year = {2002},
date = {2002-01-01},
journal = {Journal of Molecular Structure: THEOCHEM},
volume = {619},
number = {1},
pages = {195--205},
publisher = {Elsevier},
abstract = {We report here qualitative structure–activity relationship (SAR) studies for the molecular set called Tripos or Cramer steroid data set. These compounds are known to bind to corticosteroid binding globulin (CBG). In the present work we have used the electronic indices methodology (EIM). The EIM is based on Boolean relational rules exploring the concepts of local density of states and critical values for energy separation involving frontier orbitals. We have also carried out comparative principal component analysis (PCA) and hierarchical clustering analysis (HCA) studies with molecular descriptors obtained from EIM calculations. EIM, PCA and HCA correctly predict (100% accuracy) the steroid's biological activity. The present studies reinforce the universal applicability of the EIM descriptors and show that the combined use of EIM coupled to PCA can be a new efficient and powerful SAR tool.
},
keywords = {Drug Design, Electronic Structure, HCA/PCA, Neural Networks, Tripos},
pubstate = {published},
tppubtype = {article}
}
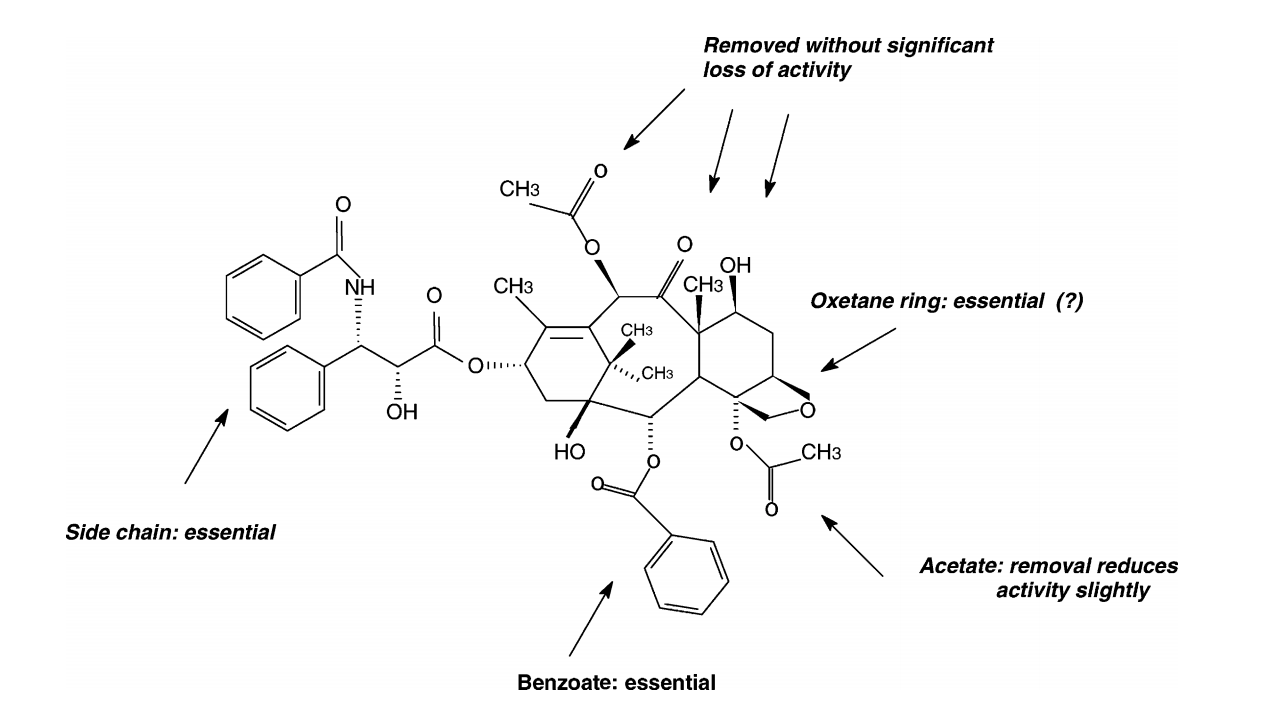
Braga, SF; Galvao, DS
A semiempirical study on the electronic structure of 10-deacetylbaccatin-III Journal Article
In: Journal of Molecular Graphics and Modelling, vol. 21, no. 1, pp. 57–70, 2002.
Abstract | Links | BibTeX | Tags: Baccatin, Drug Design, Electronic Structure, Taxol, Taxotere, Theory of Electronic Indices
@article{braga2002semiempirical,
title = {A semiempirical study on the electronic structure of 10-deacetylbaccatin-III},
author = {Braga, SF and Galvao, DS},
url = {http://www.sciencedirect.com/science/article/pii/S1093326302001213},
year = {2002},
date = {2002-01-01},
journal = {Journal of Molecular Graphics and Modelling},
volume = {21},
number = {1},
pages = {57--70},
publisher = {Elsevier},
abstract = {We performed a conformational and electronic analysis for 10-deacetylbaccatin-III (DBAC) using well-known semiempirical methods (parametric method 3 (PM3) and Zerner’s intermediate neglect of differential overlap (ZINDO)) coupled to the concepts of total and local density of states (LDOS). Our results indicate that regions presented by paclitaxel (Taxol®) as important for the biological activity can be traced out by the electronic features present in DBAC. These molecules differ only by a phenylisoserine side chain. Compared to paclitaxel, DBAC has a simpler structure in terms of molecular size and number of degrees of freedom (d.f.). This makes DBAC a good candidate for a preliminary investigation of the taxoid family. Our results question the importance of the oxetane group, which seems to be consistent with recent experimental data.
},
keywords = {Baccatin, Drug Design, Electronic Structure, Taxol, Taxotere, Theory of Electronic Indices},
pubstate = {published},
tppubtype = {article}
}
Coluci, V R; Braga, SF; Legoas, Sergio B; Galvao, Douglas S; Baughman, RH
New families of carbon nanotubes Journal Article
In: arXiv preprint cond-mat/0207085, 2002.
Abstract | Links | BibTeX | Tags: Allotropes, Electronic Structure, Graphynes, Nanotubes
@article{coluci2002new,
title = {New families of carbon nanotubes},
author = {Coluci, V R and Braga, SF and Legoas, Sergio B and Galvao, Douglas S and Baughman, RH},
url = {http://arxiv.org/abs/cond-mat/0207085},
year = {2002},
date = {2002-01-01},
journal = {arXiv preprint cond-mat/0207085},
abstract = {Fundamentally new families of carbon single walled nanotubes are proposed. These nanotubes, called graphynes, result from the elongation of covalent interconnections of graphite-based nanotubes by the introduction of yne groups. Similarly to ordinary nanotubes, arm-chair, zig-zag, and chiral graphyne nanotubes are possible. Electronic properties, predicted using tight-binding and ab initio density functional methods, show a rich variety of metallic and semiconducting behaviors.},
keywords = {Allotropes, Electronic Structure, Graphynes, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
Legoas, Sergio B; Galvao, Douglas S; Rodrigues, Varlei; Ugarte, Daniel
The Role of Carbon Contamination in Suspended Gold Nanowires Journal Article
In: MRS Proceedings, vol. 738, pp. G14–6, 2002.
Abstract | Links | BibTeX | Tags: DFT, Electronic Structure, Linear Atomic Chains, Metallic Nanowires, TEM
@article{legoas2002role,
title = {The Role of Carbon Contamination in Suspended Gold Nanowires},
author = {Legoas, Sergio B and Galvao, Douglas S and Rodrigues, Varlei and Ugarte, Daniel},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8031754&fileId=S1946427400148249},
year = {2002},
date = {2002-01-01},
journal = {MRS Proceedings},
volume = {738},
pages = {G14--6},
publisher = {Cambridge University Press},
abstract = {Metallic nanowires represent very interesting systems due to new phenomena such as quantum conductance and unexpected long interatomic distances attaining 0.3–0.5 nm. These large distances represent a challenge for physical interpretation. In this work we present experimental data from transmission electron microscopy and results from ab initio density functional calculations for suspended gold chains. We show that large distances as 0.5 nm can be easily explained by the presence of carbon atoms as contaminants, while distances ranging from 0.29 up to 0.36 nm might be explained as resulting of a mixture of clean stressed and contaminated linear chains.},
keywords = {DFT, Electronic Structure, Linear Atomic Chains, Metallic Nanowires, TEM},
pubstate = {published},
tppubtype = {article}
}
Galvao, DS; Braga, SF; Barone, PMVB; Dantas, SO
Dual-Mode Optical Molecular Switching Systems for Organic Memories Proceedings
Cambridge Univ Press, vol. 708, 2002.
Abstract | Links | BibTeX | Tags: Electronic Structure, Molecular Electronics
@proceedings{galvao2002dual,
title = {Dual-Mode Optical Molecular Switching Systems for Organic Memories},
author = {Galvao, DS and Braga, SF and Barone, PMVB and Dantas, SO},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8214320},
year = {2002},
date = {2002-01-01},
journal = {MATERIALS RESEARCH SOCIETY SYMPOSIUM PROCEEDINGS},
volume = {708},
pages = {335--340},
publisher = {Cambridge Univ Press},
abstract = {The synthesis of dual-mode optical molecular switching systems has been recently achieved. These systems were based on chiral helical-shaped alkenes in which the chirality can be reversibly modulated by light. In this work we report a theoretical study on the geometric and spectroscopic properties of these structures using the well-known semi-empirical methods PM3 (Parametric Method 3) and ZINDO/S-CI (Zerner's Intermediate Neglect of Differential Overlap -Spectroscopic - Configuration Interaction). Our results show that there are two stable conformers very close in energy for each possible molecular helicity presenting a barrier of ∼40 kcal/mol for bond rotation along the main molecular axis. Under protonation these barriers increase significantly and might explain why the protonation leads to the blocking of the switching process. We propose a scheme for the switching mechanism based on charge transfer and conformational changes during the isomer interconversion.},
keywords = {Electronic Structure, Molecular Electronics},
pubstate = {published},
tppubtype = {proceedings}
}
2001
Vendrame, R; Braga, RS; Takahata, Y; Galvao, DS
Structure--carcinogenic activity relationship studies of polycyclic aromatic hydrocarbons (PAHs) with pattern-recognition methods Journal Article
In: Journal of Molecular Structure: THEOCHEM, vol. 539, no. 1, pp. 253–265, 2001.
BibTeX | Tags:
@article{vendrame2001structure,
title = {Structure--carcinogenic activity relationship studies of polycyclic aromatic hydrocarbons (PAHs) with pattern-recognition methods},
author = {Vendrame, R and Braga, RS and Takahata, Y and Galvao, DS},
year = {2001},
date = {2001-01-01},
journal = {Journal of Molecular Structure: THEOCHEM},
volume = {539},
number = {1},
pages = {253--265},
publisher = {Elsevier},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Braga, SF; Galv ao, DS; Barone, PMVB; Dantas, SO
A theoretical investigation on the dual-mode photoswitching mechanism of some chiroptical systems Journal Article
In: The Journal of Physical Chemistry B, vol. 105, no. 35, pp. 8334–8338, 2001.
BibTeX | Tags:
@article{braga2001theoretical,
title = {A theoretical investigation on the dual-mode photoswitching mechanism of some chiroptical systems},
author = {Braga, SF and Galv~ao, DS and Barone, PMVB and Dantas, SO},
year = {2001},
date = {2001-01-01},
journal = {The Journal of Physical Chemistry B},
volume = {105},
number = {35},
pages = {8334--8338},
publisher = {American Chemical Society},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Lavarda, FC; Galv ao, DS; Laks, B
Soliton clusters inducing insulator-to-metal transition in polyacetylene: a Su--Schrieffer--Heeger model study Journal Article
In: Synthetic metals, vol. 123, no. 2, pp. 211–215, 2001.
BibTeX | Tags:
@article{lavarda2001soliton,
title = {Soliton clusters inducing insulator-to-metal transition in polyacetylene: a Su--Schrieffer--Heeger model study},
author = {Lavarda, FC and Galv~ao, DS and Laks, B},
year = {2001},
date = {2001-01-01},
journal = {Synthetic metals},
volume = {123},
number = {2},
pages = {211--215},
publisher = {Elsevier},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Galv ao, DS; Laks, B; da Silva, RR; Torres, JHS; Kopelevich, Y
High-Temperature Superconductivity in Graphite-Sulfur Composites: Theoretical Analysis Journal Article
In: MRS Proceedings, vol. 689, pp. E5–2, 2001.
BibTeX | Tags:
@article{galvao2001high,
title = {High-Temperature Superconductivity in Graphite-Sulfur Composites: Theoretical Analysis},
author = {Galv~ao, DS and Laks, B and da Silva, RR and Torres, JHS and Kopelevich, Y},
year = {2001},
date = {2001-01-01},
journal = {MRS Proceedings},
volume = {689},
pages = {E5--2},
publisher = {Cambridge University Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Dantas, S'ocrates de O; Barone, Paulo MVB; Braga, Scheila F; Galv ao, Douglas S
A dual-mode photoswitching mechanism and charge transfer on chiroptical systems—theoretical study Journal Article
In: Synthetic metals, vol. 116, no. 1, pp. 275–279, 2001.
BibTeX | Tags:
@article{dantas2001dual,
title = {A dual-mode photoswitching mechanism and charge transfer on chiroptical systems—theoretical study},
author = {Dantas, S'ocrates de O and Barone, Paulo MVB and Braga, Scheila F and Galv~ao, Douglas S},
year = {2001},
date = {2001-01-01},
journal = {Synthetic metals},
volume = {116},
number = {1},
pages = {275--279},
publisher = {Elsevier},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Galv ao, DS; Braga, SF; Barone, PMVB; Dantas, SO
Dual-Mode Optical Molecular Switching Systems for Organic Memories Journal Article
In: MRS Proceedings, vol. 708, pp. BB10–27, 2001.
BibTeX | Tags:
@article{galvao2001dual,
title = {Dual-Mode Optical Molecular Switching Systems for Organic Memories},
author = {Galv~ao, DS and Braga, SF and Barone, PMVB and Dantas, SO},
year = {2001},
date = {2001-01-01},
journal = {MRS Proceedings},
volume = {708},
pages = {BB10--27},
publisher = {Cambridge University Press},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2000
Braga, RS; Barone, PMVB; Galv ao, DS
La identificaci'on de la actividad cancer'igena de metilado y no metilado, hidrocarburos arom'aticos polic'iclicos (HAP) a trav'es de 'indices electr'onicos y topol'ogicos Journal Article
In: Brazilian Journal of Physics, vol. 30, no. 3, pp. 560–568, 2000.
BibTeX | Tags:
@article{braga2000identificacion,
title = {La identificaci'on de la actividad cancer'igena de metilado y no metilado, hidrocarburos arom'aticos polic'iclicos (HAP) a trav'es de 'indices electr'onicos y topol'ogicos},
author = {Braga, RS and Barone, PMVB and Galv~ao, DS},
year = {2000},
date = {2000-01-01},
journal = {Brazilian Journal of Physics},
volume = {30},
number = {3},
pages = {560--568},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Barone, PMVB; Braga, RS; Camilo Jr, A; Galvao, DS
Electronic indices from semi-empirical calculations to identify carcinogenic activity of polycyclic aromatic hydrocarbons Journal Article
In: Journal of Molecular Structure: THEOCHEM, vol. 505, no. 1, pp. 55–66, 2000.
BibTeX | Tags:
@article{barone2000electronic,
title = {Electronic indices from semi-empirical calculations to identify carcinogenic activity of polycyclic aromatic hydrocarbons},
author = {Barone, PMVB and Braga, RS and Camilo Jr, A and Galvao, DS},
year = {2000},
date = {2000-01-01},
journal = {Journal of Molecular Structure: THEOCHEM},
volume = {505},
number = {1},
pages = {55--66},
publisher = {Elsevier},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Braga, RS; Vendrame, Rosana; Galv ao, DS
Structure-Activity Relationship Studies of Substituted 17$alpha$-Acetoxyprogesterone Hormones Journal Article
In: Journal of chemical information and computer sciences, vol. 40, no. 6, pp. 1377–1385, 2000.
BibTeX | Tags:
@article{braga2000structure,
title = {Structure-Activity Relationship Studies of Substituted 17$alpha$-Acetoxyprogesterone Hormones},
author = {Braga, RS and Vendrame, Rosana and Galv~ao, DS},
year = {2000},
date = {2000-01-01},
journal = {Journal of chemical information and computer sciences},
volume = {40},
number = {6},
pages = {1377--1385},
publisher = {American Chemical Society},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Santo, Larissa LE; Durrant, Steven F; Rangel, Elidiane C; Galv ao, Douglas S; de Moraes, M'ario A Bica
Semi-empirical modeling of the optical gap of amorphous hydrogenated nitrogenated carbon films Journal Article
In: Journal of Vacuum Science & Technology A, vol. 18, no. 5, pp. 2466–2471, 2000.
BibTeX | Tags:
@article{santo2000semi,
title = {Semi-empirical modeling of the optical gap of amorphous hydrogenated nitrogenated carbon films},
author = {Santo, Larissa LE and Durrant, Steven F and Rangel, Elidiane C and Galv~ao, Douglas S and de Moraes, M'ario A Bica},
year = {2000},
date = {2000-01-01},
journal = {Journal of Vacuum Science & Technology A},
volume = {18},
number = {5},
pages = {2466--2471},
publisher = {AVS: Science & Technology of Materials, Interfaces, and Processing},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Braga, RS; Barone, PMVB; Galvao, DS
Identifying carcinogenic activity of methylated and non-methylated polycyclic aromatic hydrocarbons (PAHs) through electronic and topological indices Journal Article
In: Brazilian Journal of Physics, vol. 30, no. 3, pp. 560–568, 2000.
BibTeX | Tags:
@article{braga2000identifying,
title = {Identifying carcinogenic activity of methylated and non-methylated polycyclic aromatic hydrocarbons (PAHs) through electronic and topological indices},
author = {Braga, RS and Barone, PMVB and Galvao, DS},
year = {2000},
date = {2000-01-01},
journal = {Brazilian Journal of Physics},
volume = {30},
number = {3},
pages = {560--568},
publisher = {Sociedade Brasileira de F'isica},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Barone, PMVB; Braga, RS; Galv ao, DS
Identifying Carcinogenic Actividade of Methylated and non-Methylated Polycyclic Aromatic Hydrocarbons (PAHs) Through Eletronic and Topological Indices Journal Article
In: Brazilian Journal of Physics, vol. 30, pp. 560–568, 2000.
BibTeX | Tags:
@article{barone2000identifying,
title = {Identifying Carcinogenic Actividade of Methylated and non-Methylated Polycyclic Aromatic Hydrocarbons (PAHs) Through Eletronic and Topological Indices},
author = {Barone, PMVB and Braga, RS and Galv~ao, DS},
year = {2000},
date = {2000-01-01},
journal = {Brazilian Journal of Physics},
volume = {30},
pages = {560--568},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Braga, RS; Cardoso, LP; Galvao, DS; Avanci, LH
Deformation in meta-nitroaniline organic crystals through hipocycloidal anisotropy Journal Article
In: 2000.
BibTeX | Tags:
@article{braga2000deformation,
title = {Deformation in meta-nitroaniline organic crystals through hipocycloidal anisotropy},
author = {Braga, RS and Cardoso, LP and Galvao, DS and Avanci, LH},
year = {2000},
date = {2000-01-01},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ

