
Fonseca, AF; Dantas, SO; Galvao, DS; Zhang, D; Sinnott, SB
The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review) Journal Article
In: 2019.
@article{Fonseca2019d,
title = {The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review)},
author = {Fonseca, AF and Dantas, SO and Galvao, DS and Zhang, D and Sinnott, SB},
year = {2019},
date = {2019-04-03},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
AF; Dantas Fonseca, SO; Galvao
The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study Online
2019, (ArXiv preprint).
@online{Fonseca2019b,
title = {The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study},
author = {Fonseca, AF; Dantas, SO; Galvao, DS; Zhang, D; Sinnott SB},
url = {https://arxiv.org/pdf/1904.09871.pdf},
year = {2019},
date = {2019-03-22},
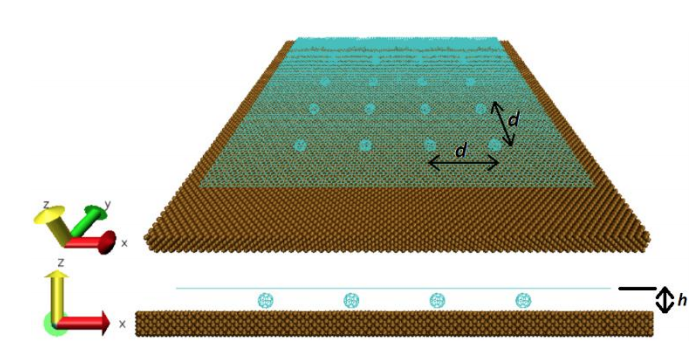
abstract = {Two experimental studies reported the spontaneous formation of amorphous and crystalline
structures of C60 intercalated between graphene and a substrate. They observed interesting
phenomena ranging from reaction between C60 molecules under graphene to graphene
sagging between the molecules and control of strain in graphene. Motivated by these works,
we performed fully atomistic reactive molecular dynamics simulations to study the formation
and thermal stability of graphene wrinkles as well as graphene attachment to and detachment
from the substrate when graphene is laid over a previously distributed array of C60 molecules
on a copper substrate at different values of temperature. As graphene compresses the C60
molecules against the substrate, and graphene attachment to the substrate between C60s
(“C60S” stands for plural of C60) depends on the height of graphene wrinkles, configurations
with both frozen and non-frozen C60s structures were investigated in order to verify the
experimental result of stable sagged graphene when the distance between C60s is about 4 nm
and height of graphene wrinkles is about 0.8 nm. Below the distance of 4 nm between C60s,
graphene becomes locally suspended and less strained. We show that this happens when C60s
are allowed to deform under the compressive action of graphene. If we keep the C60s frozen,
spontaneous “blanketing” of graphene happens only when the distance between them are
equal or above 7 nm. Both above results for the existence of stable sagged graphene for C60
distances of 4 or 7 nm are shown to agree with a mechanical model relating the rigidity of
graphene to the energy of graphene-substrate adhesion. Although the studies of intercalation
of molecules on interfaces formed by graphene-substrate are motivated by finding out ways to
control wrinkling and strain in graphene, our work reveals the shape and structure of
intercalated molecules and the role of stability and wrinkling on final structure of graphene.
In particular, this study might help the development of 2D confined nanoreactors that are
considered in literature to be the next advanced step on chemical reactions.},
note = {ArXiv preprint},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
structures of C60 intercalated between graphene and a substrate. They observed interesting
phenomena ranging from reaction between C60 molecules under graphene to graphene
sagging between the molecules and control of strain in graphene. Motivated by these works,
we performed fully atomistic reactive molecular dynamics simulations to study the formation
and thermal stability of graphene wrinkles as well as graphene attachment to and detachment
from the substrate when graphene is laid over a previously distributed array of C60 molecules
on a copper substrate at different values of temperature. As graphene compresses the C60
molecules against the substrate, and graphene attachment to the substrate between C60s
(“C60S” stands for plural of C60) depends on the height of graphene wrinkles, configurations
with both frozen and non-frozen C60s structures were investigated in order to verify the
experimental result of stable sagged graphene when the distance between C60s is about 4 nm
and height of graphene wrinkles is about 0.8 nm. Below the distance of 4 nm between C60s,
graphene becomes locally suspended and less strained. We show that this happens when C60s
are allowed to deform under the compressive action of graphene. If we keep the C60s frozen,
spontaneous “blanketing” of graphene happens only when the distance between them are
equal or above 7 nm. Both above results for the existence of stable sagged graphene for C60
distances of 4 or 7 nm are shown to agree with a mechanical model relating the rigidity of
graphene to the energy of graphene-substrate adhesion. Although the studies of intercalation
of molecules on interfaces formed by graphene-substrate are motivated by finding out ways to
control wrinkling and strain in graphene, our work reveals the shape and structure of
intercalated molecules and the role of stability and wrinkling on final structure of graphene.
In particular, this study might help the development of 2D confined nanoreactors that are
considered in literature to be the next advanced step on chemical reactions.
AF; Dantas Fonseca, SO; Galvao
The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review) Journal Article
In: 2019.
@article{Fonseca2019c,
title = {The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review)},
author = {Fonseca, AF; Dantas, SO; Galvao, DS; Zhang, D; Sinnott SB},
year = {2019},
date = {2019-03-15},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Jaques, Ygor M.; Galvao, Douglas S.
Structural Properties of Nanodroplets Impacting Graphene at High Velocities (accepted) Journal Article
In: Journal of Molecular Liquids, 2019.
@article{Jaques2019b,
title = {Structural Properties of Nanodroplets Impacting Graphene at High Velocities (accepted)},
author = {Ygor M. Jaques and Douglas S. Galvao},
year = {2019},
date = {2019-02-05},
journal = {Journal of Molecular Liquids},
abstract = {The determination of the wettability of 2D materials is an area of intensive research, as it is decisive on the applications of these systems in nanofluidics. One important part of the wetting characterization is how the spreading of droplets impacting on the surfaces occurs. However, few works address this problem for layered materials. Here, we report a fully atomistic molecular dynamics study on the dynamics of impact of water nanodroplets (100 ̊A of diameter) at high velocities (from 1 up to 15 ̊A/ps) against graphene targets. Our results show that tuning graphene wettability (through parameter changes) significantly affects the structural and dynamical aspects of the nanodroplets. We identified three ranges of velocities with distinct characteristics, from simple deposition of the droplet to spreading with rebound, and finally droplet frag- mentation. We also identify that in an intermediary velocity of 7 ̊A/ps, the pattern of spreading critically changes, due to formation of voids on droplet structure. These voids affect in a detrimental way the droplet spreading on the less hydrophilic surface, as it takes more time to the droplet recover from the spreading and to return to a semi-spherical configuration. When the velocity is increased to values larger than 11 ̊A/ps, the droplet fragments, which reveals the maximum possible spreading.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
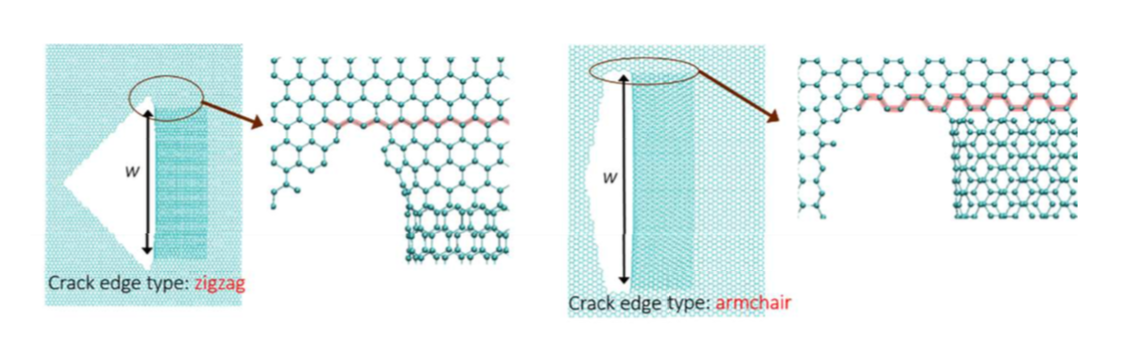
Fonseca, Alexandre F.; Galvao, Douglas S.
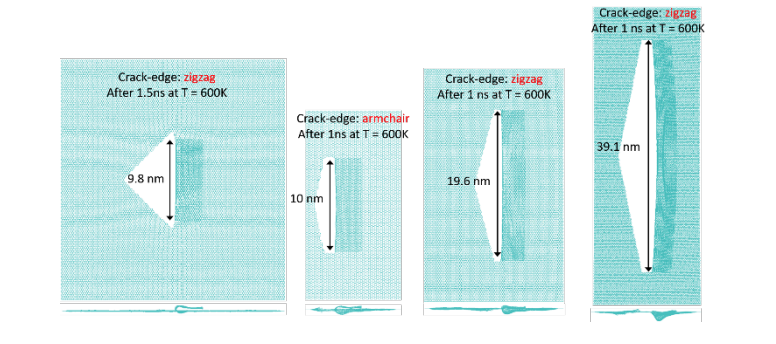
Self-tearing and self-peeling of folded graphene nanoribbons Journal Article
In: Carbon, vol. 143, pp. 230-239, 2019.
@article{Fonseca2019,
title = {Self-tearing and self-peeling of folded graphene nanoribbons},
author = {Alexandre F. Fonseca and Douglas S. Galvao},
url = {https://www.sciencedirect.com/science/article/pii/S0008622318310431},
doi = {10.1016/j.carbon.2018.11.020},
year = {2019},
date = {2019-01-05},
journal = {Carbon},
volume = {143},
pages = {230-239},
abstract = {A recent experimental study showed that an induced folded flap of graphene can spontaneously drive itself its tearing and peeling off a substrate, thus producing long, micrometer sized, regular trapezoidal-shaped folded graphene nanoribbons. As long as the size of the graphene flaps is above a threshold value, the “tug of war” between the forces of adhesion of graphene-graphene and graphene-substrate, flexural strain of folded region and carbon-carbon (C-C) covalent bonds favor the self-tearing and self-peeling off process. As the detailed information regarding the atomic scale mechanism involved in the process remains not fully understood, we carried out atomistic reactive molecular dynamics simulations to address some features of the process. We show that large thermal fluctuations can prevent the process by increasing the probability of chemical reactions between carbon dangling bonds of adjacent graphene layers. The effects of the strength of attraction between graphene and the substrate on the ribbon growth velocities at the early stages of the phenomenon were also investigated. Structures with initial armchair crack-edges were observed to form more uniform cuts than those having initial zigzag ones. Our results are of importance to help set up new experiments on this phenomenon, especially with samples with nanoscale sized cuts. },
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Bizao, Rafael A; Machado, Leonardo D; de Sousa, Jose M; Pugno, Nicola M; Galvao, Douglas S
Scale Effects on the Ballistic Penetration of Graphene Sheets Journal Article
In: Nature Scientific Reports, vol. 8, pp. 6750, 2018.
@article{Bizao2018,
title = {Scale Effects on the Ballistic Penetration of Graphene Sheets},
author = {Bizao, Rafael A and Machado, Leonardo D and de Sousa, Jose M and Pugno, Nicola M and Galvao, Douglas S},
url = {https://www.nature.com/articles/s41598-018-25050-2},
doi = {doi:10.1038/s41598-018-25050-2},
year = {2018},
date = {2018-04-30},
journal = {Nature Scientific Reports},
volume = {8},
pages = {6750},
abstract = {Carbon nanostructures are promising ballistic protection materials, due to their low density and excellent mechanical properties. Recent experimental and computational investigations on the behavior of graphene under impact conditions revealed exceptional energy absorption properties as well. However, the reported numerical and experimental values differ by an order of magnitude. In this work, we combined numerical and analytical modeling to address this issue. In the numerical part, we employed reactive molecular dynamics to carry out ballistic tests on single, double, and triple-layered graphene sheets. We used velocity values within the range tested in experiments. Our numerical and the experimental results were used to determine parameters for a scaling law. We find that the specific penetration energy decreases as the number of layers (N) increases, from ∼15 MJ/kg for N = 1 to ∼0.9 MJ/kg for N = 350, for an impact velocity of 900 m/s. These values are in good agreement with simulations and experiments, within the entire range of N values for which data is presently available. Scale effects explain the apparent discrepancy between simulations and experiments.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Ygor M.; Galvao Jaques, Douglas S.
Structural Properties of Nanodroplets Impacting Graphene at High Velocities Online
2018, (Preprint ArXiv:1804.07784).
@online{Jaques2018d,
title = {Structural Properties of Nanodroplets Impacting Graphene at High Velocities},
author = {Jaques, Ygor M.; Galvao, Douglas S.},
url = {https://arxiv.org/abs/1804.07784},
year = {2018},
date = {2018-04-24},
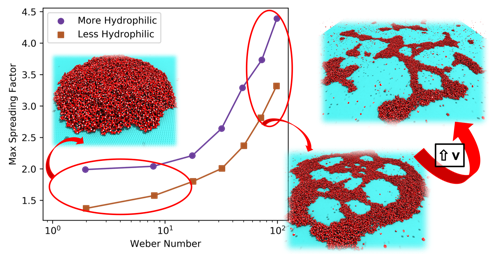
abstract = {We report here a fully atomistic molecular dynamics study on the dynamics of impact of water
nanodroplets (50, 100 and 120 Å of diameter) at high velocity (from 100 up to 1000 m/s) against
graphene targets. Our results show that tuning graphene wettability (through parameter changes)
significantly affects the structural and dynamical aspects of the nanodroplets. We identified three
ranges of velocities with distinct characteristics, from simple deposition of the droplet to
spreading with rebound and finally fragmentation. At Weber numbers lower than 10, the droplets
maintain a steady spreading factor independent of size. After this threshold value, the spread
rapidly grows with increasing Weber numbers. A more hydrophilic graphene surface increases
the spreading values, due to stronger solid-liquid interactions. Nevertheless, droplet size also
influences the fragmentation threshold, as an increased number of molecules make it easier for
the whole droplet overcomes the surface repulsion. },
note = {Preprint ArXiv:1804.07784},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
nanodroplets (50, 100 and 120 Å of diameter) at high velocity (from 100 up to 1000 m/s) against
graphene targets. Our results show that tuning graphene wettability (through parameter changes)
significantly affects the structural and dynamical aspects of the nanodroplets. We identified three
ranges of velocities with distinct characteristics, from simple deposition of the droplet to
spreading with rebound and finally fragmentation. At Weber numbers lower than 10, the droplets
maintain a steady spreading factor independent of size. After this threshold value, the spread
rapidly grows with increasing Weber numbers. A more hydrophilic graphene surface increases
the spreading values, due to stronger solid-liquid interactions. Nevertheless, droplet size also
influences the fragmentation threshold, as an increased number of molecules make it easier for
the whole droplet overcomes the surface repulsion.
Devi, M. Manolata; Dolai, N.; S, S. Sreehala; Jaques, Y. M.; Galvao, Douglas S.; C.S.Tiwary,; Sharma, Sudhanshu; Biswas, Krishanu
Morphology Controlled Graphene-Alloy Nanoparticles Hybrids with Tunable Catalytic Activity Journal Article
In: Nanoscale, vol. 10, pp. 8840-8850, 2018.
@article{Devi2018b,
title = {Morphology Controlled Graphene-Alloy Nanoparticles Hybrids with Tunable Catalytic Activity},
author = {M. Manolata Devi and N. Dolai and S. Sreehala S and Y. M. Jaques and Douglas S. Galvao and C.S.Tiwary and Sudhanshu Sharma and Krishanu Biswas},
url = {pubs.rsc.org/en/content/articlehtml/2018/nr/c7nr09688g},
doi = {10.1039/C7NR09688G},
year = {2018},
date = {2018-04-07},
journal = {Nanoscale},
volume = {10},
pages = {8840-8850},
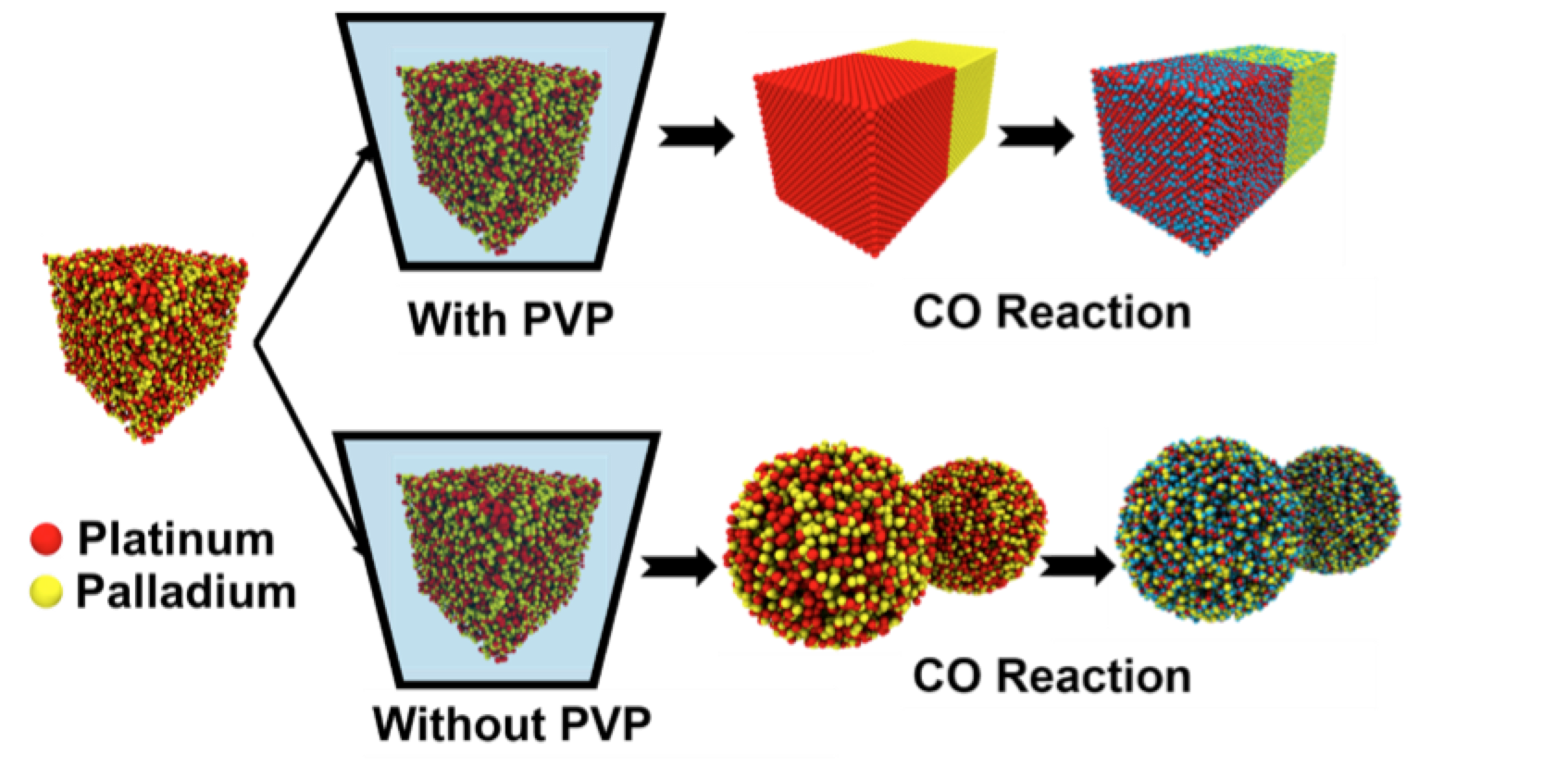
abstract = {Selective oxidation of CO to CO2 using metallic or alloy nanoparticles as catalysts can solve two major problems of energy requirements and environmental pollution. Achieving 100% conversion efficiency at a lower temperature is a very important goal. This requires sustained efforts to design and develop novel supported catalysts containing alloy nanoparticles. In this regard, the decoration of nanoalloys with graphene, as a support for the catalyst, can provide a novel structure due to the synergic effect of the nanoalloys and graphene. Here, we demonstrate the effect of nano-PdPt (Palladium–Platinum) alloys having different morphologies on the catalytic efficiency for the selective oxidation of CO. Efforts were made to prepare different morphologies of PdPt alloy nanoparticles with the advantage of tuning the capping agent (PVP – polyvinyl pyrollidone) and decorating them on graphene sheets via the wet-chemical route. The catalytic activity of the G-PdPt hybrids with an urchin-like morphology has been found to be superior (higher % conversion at 135 °C lower) to that with a nanoflower morphology. The above experimental observations are further supported by molecular dynamics (MD) simulations.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
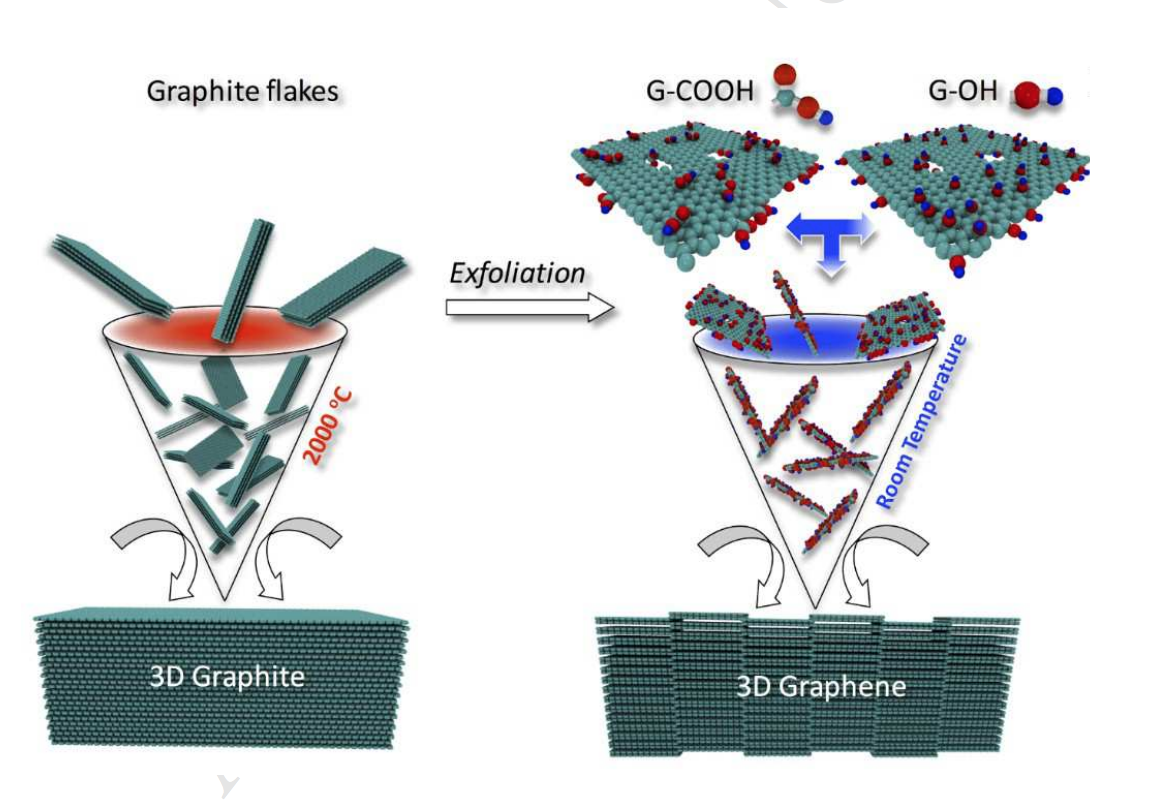
Kabbani, Mohamad A.; Kochat, Vidya; Bhowmick, Sanjit; Soto, Matias; Som, Anirban; Krishnadas, K. R.; Woellner, Cristiano F.; Jaques, Ygor M.; Barrera, Enrique V.; Asif, Syed; Vajtai, Robert; Pradeep, Thalappil; Galvão, Douglas S.; Kabbani, Ahmad T.; Tiwary, Chandra Sekhar; Ajayan, Pulickel M.
Consolidation of Functionalized Graphene at Ambient Temperature via Mechano-chemistry Journal Article
In: Carbon, vol. 134, no. 8, pp. 491-499, 2018.
@article{Kabbani2018,
title = {Consolidation of Functionalized Graphene at Ambient Temperature via Mechano-chemistry},
author = {Mohamad A. Kabbani and Vidya Kochat and Sanjit Bhowmick and Matias Soto and Anirban Som and K.R. Krishnadas and Cristiano F. Woellner and Ygor M. Jaques and Enrique V. Barrera and Syed Asif and Robert Vajtai and Thalappil Pradeep and Douglas S. Galvão and Ahmad T. Kabbani and Chandra Sekhar Tiwary and Pulickel M. Ajayan},
url = {https://www.sciencedirect.com/science/article/pii/S0008622318302987?dgcid=raven_sd_aip_email},
doi = {DOI:10.1016/j.carbon.2018.03.049},
year = {2018},
date = {2018-03-22},
journal = {Carbon},
volume = {134},
number = {8},
pages = {491-499},
abstract = {Graphitic solids are typically produced via high temperature and energy consuming
processing (e.g. sintering) of carbon particles. Here, we demonstrate the mechano-chemical
assembly of functionalized graphene layers into 3D graphitic solids via room temperature and
low energy consuming processing. The chemical functional groups on graphene layers are
interconnected at room temperature under pressure leading to porous three-dimensional
structures with tunable mechanical and electrical properties. The formation of mechanochemistry
induced atomic scale junctions and their impact on mechanical properties of
graphene assembled carbon materials are demonstrated through nano-indentation experiments
and confirmed using DFT and molecular dynamics simulations. The results show room
temperature consolidation routes of graphene layers into bulk carbon solids.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
processing (e.g. sintering) of carbon particles. Here, we demonstrate the mechano-chemical
assembly of functionalized graphene layers into 3D graphitic solids via room temperature and
low energy consuming processing. The chemical functional groups on graphene layers are
interconnected at room temperature under pressure leading to porous three-dimensional
structures with tunable mechanical and electrical properties. The formation of mechanochemistry
induced atomic scale junctions and their impact on mechanical properties of
graphene assembled carbon materials are demonstrated through nano-indentation experiments
and confirmed using DFT and molecular dynamics simulations. The results show room
temperature consolidation routes of graphene layers into bulk carbon solids.
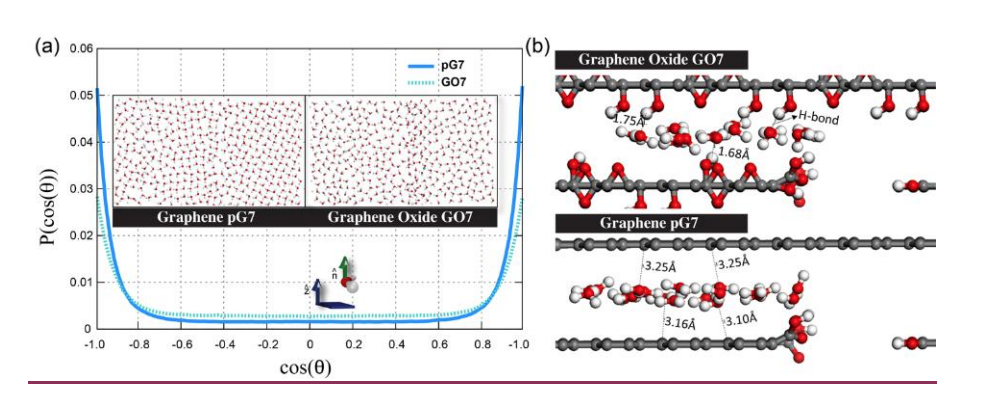
Borges, Daiane Damasceno; Woellner, Cristiano F.; Autreto, Pedro A. S.; Galvao, Douglas S.
Water/alcohol separation via layered oxide graphene membranes Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 109-114, 2018.
@article{Borges2018d,
title = {Water/alcohol separation via layered oxide graphene membranes},
author = {Daiane Damasceno Borges and Cristiano F. Woellner and Pedro A. S. Autreto and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/wateralcohol-separation-in-graphene-oxide-membranes-insights-from-molecular-dynamics-and-monte-carlo-simulations/C61C66FF48D35EB2DB3408ACCE96C41A},
doi = { https://doi.org/10.1557/adv.2018.192},
year = {2018},
date = {2018-02-13},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = {109-114},
abstract = {Graphene-based membranes have been investigated as promising candidates for water filtration and gas separation applications. Experimental evidences have shown that graphene oxide can be impermeable to liquids, vapors and gases, while allowing a fast permeation of water molecules. This phenomenon has been attributed to the formation of a network of nano capillaries that allow nearly frictionless water flow while blocking other molecules by steric hindrance effects. It is supposed that water molecules are transported through the percolated two-dimensional channels formed between graphene-based sheets. Although these channels allow fast water permeation in such materials, the flow rates are strongly dependent on how the membranes are fabricated. Also, some fundamental issues regarding the nanoscale mechanisms of water permeation are still not fully understood and their interpretation remains controversial. In this work, we have investigated the dynamics of water permeation through pristine graphene and graphene oxide model membranes that have strong impact on water/alcohol separation. We have carried out fully atomistic classical molecular dynamics simulations of systems composed of multiple layered graphene-based sheets into contact with a pure water reservoir under controlled thermodynamics conditions (e. g., by varying temperature and pressure values). We have systematically analysed how the transport dynamics of the confined nanofluids depend on the interlayer distances and the role of the oxide functional groups. Our results show the water flux is much more effective for graphene than for graphene oxide membranes. These results can be attributed to the H-bonds formation between oxide functional groups and water, which traps the water molecules and precludes ultrafast water transport through the nanochannels.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
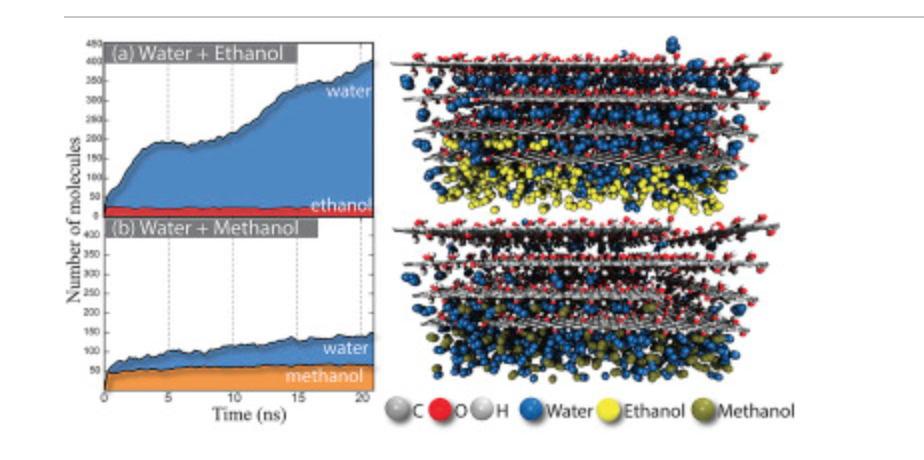
Cristiano F Woellner Daiane Damasceno Borges, Pedro AS Autreto
Insights on the mechanism of water-alcohol separation in multilayer graphene oxide membranes: entropic versus enthalpic factors Journal Article
In: Carbon, vol. 127, pp. 280-286, 2018.
@article{Borges2018,
title = {Insights on the mechanism of water-alcohol separation in multilayer graphene oxide membranes: entropic versus enthalpic factors},
author = {Daiane Damasceno Borges, Cristiano F Woellner, Pedro AS Autreto, Douglas S Galvao},
url = {https://www.sciencedirect.com/science/article/pii/S000862231731134X},
doi = {https://doi.org/10.1016/j.carbon.2017.11.020},
year = {2018},
date = {2018-02-01},
journal = {Carbon},
volume = {127},
pages = {280-286},
abstract = {xperimental evidence has shown that graphene oxide (GO) can be impermeable to liquids, vapors and gases, while it allows a fast permeation of water molecules. Theoretical studies to understand the filtration mechanisms come mostly from water desalination, while very few works have been dedicated to alcohol dehydration. In this work, we have investigated the molecular level mechanism underlying the alcohol/water separation inside GO membranes. A series of Molecular Dynamics and Grand-Canonical Monte Carlo simulations were carried out to probe the ethanol/water and methanol/water separation through GO membranes composed of multiple layered graphene-based films with different interlayer distance values and number of oxygen-containing functional groups. Our results show that the size exclusion and membrane affinities are not sufficient to explain the selectivity. Besides that, the favorable water molecular arrangement inside GO 2D-channels forming a robust H-bond network and the fast water permeation are crucial for an effective separation mechanism. In other words, the separation phenomenon is not only governed by membrane affinities (enthalpic mechanisms) but mainly by the geometry and size factors (entropic mechanisms). Our findings are consistent with the available experimental data and contribute to clarify important aspects of the separation behavior of confined alcohol/water in GO membranes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Fonseca, Alexandre F.; Galvao, Douglas S.
Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation Journal Article
In: MRS Advances, vol. 3, no. 8-9, pp. 460-465, 2018.
@article{Fonseca2018,
title = {Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation},
author = {Alexandre F. Fonseca and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/selfdriven-graphene-tearing-and-peeling-a-fully-atomistic-molecular-dynamics-investigation/BFC76FC4479AA617E16FA6AC7AB4D487},
doi = {https://doi.org/10.1557/adv.2018.120},
year = {2018},
date = {2018-01-30},
journal = {MRS Advances},
volume = {3},
number = {8-9},
pages = {460-465},
abstract = {In spite of years of intense research, graphene continues to produce surprising results. Recently, it was experimentally observed that under certain conditions graphene can self-drive its tearing and peeling from substrates. This process can generate long, micrometer sized, folded nanoribbons without the action of any external forces. Also, during this cracking-like propagation process, the width of the graphene folded ribbon continuously decreases and the process only stops when the width reaches about few hundreds nanometers in size. It is believed that interplay between the strain energy of folded regions, breaking of carbon-carbon covalent bonds, and adhesion of graphene-graphene and graphene-substrate are the most fundamental features of this process, although the detailed mechanisms at atomic scale remain unclear. In order to gain further insights on these processes we carried out fully atomistic reactive molecular dynamics simulations using the AIREBO potential as available in the LAMMPS computational package. Although the reported tearing/peeling experimental observations were only to micrometer sized structures, our results showed that they could also occur at nanometer scale. Our preliminary results suggest that the graphene tearing/peeling process originates from thermal energy fluctuations that results in broken bonds, followed by strain release that creates a local elastic wave that can either reinforce the process, similar to a whip cracking propagation, or undermine it by producing carbon dangling bonds that evolve to the formation of bonds between the two layers of graphene. As the process continues in time and the folded graphene decreases in width, the carbon-carbon bonds at the ribbon edge and interlayer bonds get less stressed, thermal fluctuations become unable to break them and the process stops.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Fonseca, Alexandre F.; Galvao, Douglas S.
Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation Online
2018, (preprint arXiv:1801.05354).
@online{Fonseca2018b,
title = {Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation},
author = {Alexandre F. Fonseca and Douglas S. Galvao
},
url = {https://arxiv.org/abs/1801.05354},
year = {2018},
date = {2018-01-17},
abstract = {In spite of years of intense research, graphene continues to produce surprising results. Recently, it was experimentally observed that under certain conditions graphene can self-drive its tearing and peeling from substrates. This process can generate long, micrometer sized, folded nanoribbons without the action of any external forces. Also, during this cracking-like propagation process, the width of the graphene folded ribbon continuously decreases and the process only stops when the width reaches about few hundreds nanometers in size. It is believed that interplay between the strain energy of folded regions, breaking of carbon-carbon covalent bonds, and adhesion of graphene-graphene and graphene-substrate are the most fundamental features of this process, although the detailed mechanisms at atomic scale remain unclear. In order to gain further insights on these processes we carried out fully atomistic reactive molecular dynamics simulations using the AIREBO potential as available in the LAMMPS computational package. Although the reported tearing/peeling experimental observations were only to micrometer sized structures, our results showed that they could also occur at nanometer scale. Our preliminary results suggest that the graphene tearing/peeling process originates from thermal energy fluctuations that results in broken bonds, followed by strain release that creates a local elastic wave that can either reinforce the process, similar to a whip cracking propagation, or undermine it by producing carbon dangling bonds that evolve to the formation of bonds between the two layers of graphene. As the process continues in time and the folded graphene decreases in width, the carbon-carbon bonds at the ribbon edge and interlayer bonds get less stressed, thermal fluctuations become unable to break them and the process stops.},
note = {preprint arXiv:1801.05354},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Oliveira, Eliezer Fernando; Santos, Ricardo Paupitz; da Silva Autreto, Pedro Alves; Stanislav Moshkalev,; Galvao, Douglas Soares
Improving Graphene-metal Contacts: Thermal Induced Polishing Online
2018, (preprint ArXiv:1801.04785).
@online{Oliveira2018d,
title = {Improving Graphene-metal Contacts: Thermal Induced Polishing},
author = {Eliezer Fernando Oliveira and Ricardo Paupitz Santos and Pedro Alves da Silva Autreto and Stanislav Moshkalev, and Douglas Soares Galvao},
url = {https://arxiv.org/abs/1801.04785},
year = {2018},
date = {2018-01-15},
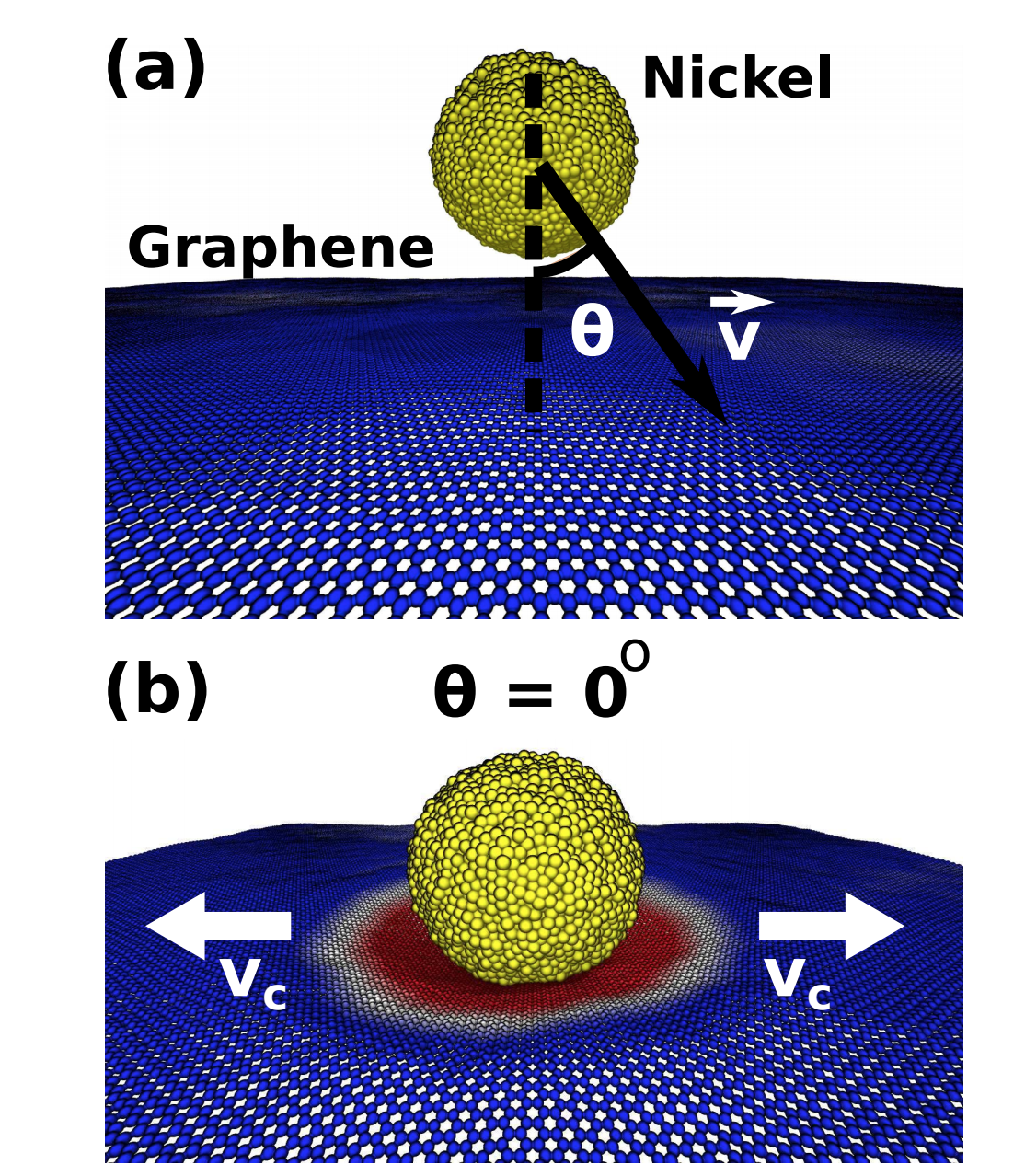
abstract = {Graphene is a very promising material for nanoelectronics applications due to its unique and remarkable electronic and thermal properties. However, when deposited on metallic electrodes the overall thermal conductivity is significantly decreased. This phenomenon has been attributed to the mismatch between the interfaces and contact thermal resistance. Experimentally, one way to improve the graphene/metal contact is thorough high-temperature annealing, but the detailed mechanisms behind these processes remain unclear. In order to address these questions, we carried out fully atomistic reactive molecular dynamics simulations using the ReaxFF force field to investigate the interactions between multi-layer graphene and metallic electrodes (nickel) under (thermal) annealing. Our results show that the annealing induces an upward-downward movement of the graphene layers, causing a pile- driver-like effect over the metallic surface. This graphene induced movements cause a planarization (thermal polishing-like effect) of the metallic surface, which results in the increase of the effective graphene/metal contact area. This can also explain the experimentally observed improvements of the thermal and electric conductivities.},
note = {preprint ArXiv:1801.04785},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Oliveira, Eliezer Fernando; Paupitz, Ricardo; da Silva Autreto, Pedro Alves; Moshkalev, Stanislav; Galvao, Douglas Soares
Improving Graphene-metal Contacts: Thermal Induced Polishing Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 73-78, 2018.
@article{Oliveira2018c,
title = {Improving Graphene-metal Contacts: Thermal Induced Polishing },
author = {Eliezer Fernando Oliveira and Ricardo Paupitz and Pedro Alves da Silva Autreto and Stanislav Moshkalev and Douglas Soares Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/improving-graphenemetal-contacts-thermal-induced-polishing/AC01C4996B90B0EE5E03220604071D12},
doi = {https://doi.org/10.1557/adv.2018.66},
year = {2018},
date = {2018-01-01},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = {73-78},
abstract = {Graphene is a very promising material for nanoelectronics applications due to its unique and remarkable electronic and thermal properties. However, when deposited on metallic electrodes the overall thermal conductivity is significantly decreased. This phenomenon has been attributed to the mismatch between the interfaces and contact thermal resistance. Experimentally, one way to improve the graphene/metal contact is thorough high-temperature annealing, but the detailed mechanisms behind these processes remain unclear. In order to address these questions, we carried out fully atomistic reactive molecular dynamics simulations using the ReaxFF force field to investigate the interactions between multi-layer graphene and metallic electrodes (nickel) under (thermal) annealing. Our results show that the annealing induces an upward-downward movement of the graphene layers, causing a pile-driver-like effect over the metallic surface. This graphene induced movements cause a planarization (thermal polishing-like effect) of the metallic surface, which results in the increase of the effective graphene/metal contact area. This can also explain the experimentally observed improvements of the thermal and electric conductivities.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Miyazaki, Celina M; Maria, Marco AE; Borges, Daiane Damasceno; Woellner, Cristiano F; Brunetto, Gustavo; Fonseca, Alexandre F; Constantino, Carlos JL; Pereira-da-Silva, Marcelo A; de Siervo, Abner; Galvao, Douglas S; Riul Jr., Antonio
2017, (preprint arXiv:1702.00250).
@online{Miyazaki2017,
title = {Synthesis, characterization and computational simulation of graphene nanoplatelets stabilized in poly (styrene sulfonate) sodium salt},
author = {Miyazaki, Celina M and Maria, Marco AE and Borges, Daiane Damasceno and Woellner, Cristiano F and Brunetto, Gustavo and Fonseca, Alexandre F and Constantino, Carlos JL and Pereira-da-Silva, Marcelo A and de Siervo, Abner and Galvao, Douglas S and Riul Jr., Antonio},
url = {https://arxiv.org/abs/1705.10673},
year = {2017},
date = {2017-05-30},
abstract = {The production of large area interfaces and the use of scalable methods to build-up designed nanostructures generating advanced functional properties are of high interest for many materials science applications. Nevertheless, large area coverage remains a major problem for pristine graphene and here we present a hybrid, composite graphene-like material soluble in water, which can be exploited in many areas, such as energy storage, electrodes fabrication, selective membranes and biosensing. Graphene oxide (GO) was produced by the traditional Hummers method being further reduced in the presence of poly(styrene sulfonate) sodium salt (PSS), thus creating stable reduced graphene oxide (rGO) nanoplateles wrapped by PSS (GPSS). Molecular dynamics simulations were carried out of further clarify the interactions between PSS molecules and rGO nanoplatelets, with calculations supported by FTIR analysis. The intermolecular forces between rGO nanoplatelets and PSS lead to the formation of a hybrid material (GPSS) stabilized by van der Waals forces, allowing the fabrication of high quality layer-by-layer (LbL) films with polyalillamine hydrochloride (PAH). Raman and electrical characterizations corroborated the successful modifications in the electronic structures from GO to GPSS after the chemical treatment, resulting in (PAH/GPSS) LbL films four orders of magnitude more conductive than (PAH/GO).
},
note = {preprint arXiv:1702.00250},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Bizao, Rafael A; Botari, Tiago; Perim, Eric; Pugno, Nicola M; Galvao, Douglas S
Mechanical properties and fracture patterns of graphene (graphitic) nanowiggles Journal Article
In: Carbon, vol. 119, pp. 431-437, 2017, (See also ArxIv version: https://arxiv.org/abs/1702.01100).
@article{Bizao2017b,
title = {Mechanical properties and fracture patterns of graphene (graphitic) nanowiggles},
author = {Bizao, Rafael A and Botari, Tiago and Perim, Eric and Pugno, Nicola M and Galvao, Douglas S},
url = {http://www.sciencedirect.com/science/article/pii/S0008622317303743},
doi = {10.1016/j.carbon.2017.04.018},
year = {2017},
date = {2017-04-14},
journal = {Carbon},
volume = {119},
pages = {431-437},
abstract = {Graphene nanowiggles (GNW) are graphene-based nanostructures obtained by making alternated regular cuts in pristine graphene nanoribbons. GNW were recently synthesized and it was demonstrated that they exhibit tunable electronic and magnetic properties by just varying their shape. Here, we have investigated the mechanical properties and fracture patterns of a large number of GNW of different shapes and sizes using fully atomistic reactive molecular dynamics simulations. Our results show that the GNW mechanical properties are strongly dependent on its shape and size and, as a general trend narrow sheets have larger ultimate strength and Young's modulus than wide ones. The estimated Young's modulus values were found to be in a range of ≈100−1000 GPa and the ultimate strength in a range of ≈20−110 GPa, depending on GNW shape. Also, super-ductile behavior under strain was observed for some structures.},
note = {See also ArxIv version: https://arxiv.org/abs/1702.01100},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Splugues, Vinicius; da Silva Autreto, Pedro Alves; Galvao, Douglas S
Hydrogenation Dynamics of Biphenylene Carbon (Graphenylene) Membranes Journal Article
In: MRS Advances, vol. 2017, pp. 1-6, 2017.
@article{Splugues2017,
title = {Hydrogenation Dynamics of Biphenylene Carbon (Graphenylene) Membranes},
author = {Splugues, Vinicius and da Silva Autreto, Pedro Alves and Galvao, Douglas S},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/hydrogenation-dynamics-of-biphenylene-carbon-graphenylene-membranes/139DB900D41560D64F352A31CE219D3A},
doi = {10.1557/adv.2017.239},
year = {2017},
date = {2017-02-28},
journal = {MRS Advances},
volume = {2017},
pages = {1-6},
abstract = {The advent of graphene created a revolution in materials science. Because of this there is a renewed interest in other carbon-based structures. Graphene is the ultimate (just one atom thick) membrane. It has been proposed that graphene can work as impermeable membrane to standard gases, such argon and helium. Graphene-like porous membranes, but presenting larger porosity and potential selectivity would have many technological applications. Biphenylene carbon (BPC), sometimes called graphenylene, is one of these structures. BPC is a porous two-dimensional (planar) allotrope carbon, with its pores resembling typical sieve cavities and/or some kind of zeolites. In this work, we have investigated the hydrogenation dynamics of BPC membranes under different conditions (hydrogenation plasma density, temperature, etc.). We have carried out an extensive study through fully atomistic molecular dynamics (MD) simulations using the reactive force field ReaxFF, as implemented in the well-known Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) code. Our results show that the BPC hydrogenation processes exhibit very complex patterns and the formation of correlated domains (hydrogenated islands) observed in the case of graphene hydrogenation was also observed here. MD results also show that under hydrogenation BPC structure undergoes a change in its topology, the pores undergoing structural transformations and extensive hydrogenation can produce significant structural damages, with the formation of large defective areas and large structural holes, leading to structural collapse.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Bizao, Rafael A; Botari, Tiago; Perim, Eric; Pugno, Nicola M; Galvao, Douglas S
Mechanical Properties and Fracture Patterns of Graphene (Graphitic) Nanowiggles Online
2017, (preprint arXiv:1702.01100).
@online{Bizao2017,
title = {Mechanical Properties and Fracture Patterns of Graphene (Graphitic) Nanowiggles},
author = {Bizao, Rafael A and Botari, Tiago and Perim, Eric and Pugno, Nicola M and Galvao, Douglas S},
url = {https://arxiv.org/pdf/1702.01100.pdf},
year = {2017},
date = {2017-02-03},
abstract = {Graphene nanowiggles (GNW) are graphene-based nanostructures
obtained by making alternated regular cuts in pristine graphene nanoribbons.
GNW were recently synthesized and it was demonstrated that
they exhibit tunable electronic and magnetic properties by just varying
their shape. Here, we have investigated the mechanical properties and
fracture patterns of a large number of GNW of different shapes and
sizes using fully atomistic reactive molecular dynamics simulations.
Our results show that the GNW mechanical properties are strongly
dependent on its shape and size and, as a general trend narrow sheets
have larger ultimate strength and Young’s modulus than wide ones.
The estimated Young’s modulus values were found to be in a range of
≈ 100 − 1000 GPa and the ultimate strength in a range of ≈ 20 − 110
GPa, depending on GNW shape. Also, super-ductile behaviour under
strain was observed for some structures.},
note = {preprint arXiv:1702.01100},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
obtained by making alternated regular cuts in pristine graphene nanoribbons.
GNW were recently synthesized and it was demonstrated that
they exhibit tunable electronic and magnetic properties by just varying
their shape. Here, we have investigated the mechanical properties and
fracture patterns of a large number of GNW of different shapes and
sizes using fully atomistic reactive molecular dynamics simulations.
Our results show that the GNW mechanical properties are strongly
dependent on its shape and size and, as a general trend narrow sheets
have larger ultimate strength and Young’s modulus than wide ones.
The estimated Young’s modulus values were found to be in a range of
≈ 100 − 1000 GPa and the ultimate strength in a range of ≈ 20 − 110
GPa, depending on GNW shape. Also, super-ductile behaviour under
strain was observed for some structures.
Borges, Daiane D; Woellner, Cristiano F; Autreto, Pedro AS; Galvao, Douglas S
2017, (preprint arXiv:1702.00250).
@online{Borges2017,
title = {Water Permeation through Layered Graphene-based Membranes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Borges, Daiane D and Woellner, Cristiano F and Autreto, Pedro AS and Galvao, Douglas S},
url = {https://arxiv.org/abs/1702.00250},
year = {2017},
date = {2017-02-01},
abstract = {Graphene-based membranes have been investigated as promising candidates for water
filtration and gas separation applications. Experimental evidences have shown that graphene
oxide can be impermeable to liquids, vapors and gases, while allowing a fast permeation of water
molecules. This phenomenon has been attributed to the formation of a network of nano
capillaries that allow nearly frictionless water flow while blocking other molecules by steric
hindrance effects. It is supposed that water molecules are transported through the percolated twodimensional
channels formed between graphene-based sheets. Although these channels allow
fast water permeation in such materials, the flow rates are strongly dependent on how the
membranes are fabricated. Also, some fundamental issues regarding the nanoscale mechanisms
of water permeation are still not fully understood and their interpretation remains controversial.
In this work, we have investigated the dynamics of water permeation through pristine graphene
and graphene oxide model membranes. We have carried out fully atomistic classical molecular
dynamics simulations of systems composed of multiple layered graphene-based sheets into
contact with a water reservoir under controlled thermodynamics conditions (e. g., by varying
temperature and pressure values). We have systematically analyzed how the transport dynamics
of the confined nanofluids depend on the interlayer distances and the role of the oxide functional
groups. Our results show the water flux is much more effective for graphene than for graphene
oxide membranes. These results are attributed to the H-bonds formation between oxide
functional groups and water, which traps the water molecules and precludes ultrafast water
transport through the nanochannels.},
note = {preprint arXiv:1702.00250},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
filtration and gas separation applications. Experimental evidences have shown that graphene
oxide can be impermeable to liquids, vapors and gases, while allowing a fast permeation of water
molecules. This phenomenon has been attributed to the formation of a network of nano
capillaries that allow nearly frictionless water flow while blocking other molecules by steric
hindrance effects. It is supposed that water molecules are transported through the percolated twodimensional
channels formed between graphene-based sheets. Although these channels allow
fast water permeation in such materials, the flow rates are strongly dependent on how the
membranes are fabricated. Also, some fundamental issues regarding the nanoscale mechanisms
of water permeation are still not fully understood and their interpretation remains controversial.
In this work, we have investigated the dynamics of water permeation through pristine graphene
and graphene oxide model membranes. We have carried out fully atomistic classical molecular
dynamics simulations of systems composed of multiple layered graphene-based sheets into
contact with a water reservoir under controlled thermodynamics conditions (e. g., by varying
temperature and pressure values). We have systematically analyzed how the transport dynamics
of the confined nanofluids depend on the interlayer distances and the role of the oxide functional
groups. Our results show the water flux is much more effective for graphene than for graphene
oxide membranes. These results are attributed to the H-bonds formation between oxide
functional groups and water, which traps the water molecules and precludes ultrafast water
transport through the nanochannels.
2019
Fonseca, AF; Dantas, SO; Galvao, DS; Zhang, D; Sinnott, SB
The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review) Journal Article
In: 2019.
BibTeX | Tags: C60, Graphene, Molecular Dynamics
@article{Fonseca2019d,
title = {The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review)},
author = {Fonseca, AF and Dantas, SO and Galvao, DS and Zhang, D and Sinnott, SB},
year = {2019},
date = {2019-04-03},
keywords = {C60, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
AF; Dantas Fonseca, SO; Galvao
The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study Online
2019, (ArXiv preprint).
Abstract | Links | BibTeX | Tags: C60, Graphene, Molecular Dynamics
@online{Fonseca2019b,
title = {The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study},
author = {Fonseca, AF; Dantas, SO; Galvao, DS; Zhang, D; Sinnott SB},
url = {https://arxiv.org/pdf/1904.09871.pdf},
year = {2019},
date = {2019-03-22},
abstract = {Two experimental studies reported the spontaneous formation of amorphous and crystalline
structures of C60 intercalated between graphene and a substrate. They observed interesting
phenomena ranging from reaction between C60 molecules under graphene to graphene
sagging between the molecules and control of strain in graphene. Motivated by these works,
we performed fully atomistic reactive molecular dynamics simulations to study the formation
and thermal stability of graphene wrinkles as well as graphene attachment to and detachment
from the substrate when graphene is laid over a previously distributed array of C60 molecules
on a copper substrate at different values of temperature. As graphene compresses the C60
molecules against the substrate, and graphene attachment to the substrate between C60s
(“C60S” stands for plural of C60) depends on the height of graphene wrinkles, configurations
with both frozen and non-frozen C60s structures were investigated in order to verify the
experimental result of stable sagged graphene when the distance between C60s is about 4 nm
and height of graphene wrinkles is about 0.8 nm. Below the distance of 4 nm between C60s,
graphene becomes locally suspended and less strained. We show that this happens when C60s
are allowed to deform under the compressive action of graphene. If we keep the C60s frozen,
spontaneous “blanketing” of graphene happens only when the distance between them are
equal or above 7 nm. Both above results for the existence of stable sagged graphene for C60
distances of 4 or 7 nm are shown to agree with a mechanical model relating the rigidity of
graphene to the energy of graphene-substrate adhesion. Although the studies of intercalation
of molecules on interfaces formed by graphene-substrate are motivated by finding out ways to
control wrinkling and strain in graphene, our work reveals the shape and structure of
intercalated molecules and the role of stability and wrinkling on final structure of graphene.
In particular, this study might help the development of 2D confined nanoreactors that are
considered in literature to be the next advanced step on chemical reactions.},
note = {ArXiv preprint},
keywords = {C60, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}
structures of C60 intercalated between graphene and a substrate. They observed interesting
phenomena ranging from reaction between C60 molecules under graphene to graphene
sagging between the molecules and control of strain in graphene. Motivated by these works,
we performed fully atomistic reactive molecular dynamics simulations to study the formation
and thermal stability of graphene wrinkles as well as graphene attachment to and detachment
from the substrate when graphene is laid over a previously distributed array of C60 molecules
on a copper substrate at different values of temperature. As graphene compresses the C60
molecules against the substrate, and graphene attachment to the substrate between C60s
(“C60S” stands for plural of C60) depends on the height of graphene wrinkles, configurations
with both frozen and non-frozen C60s structures were investigated in order to verify the
experimental result of stable sagged graphene when the distance between C60s is about 4 nm
and height of graphene wrinkles is about 0.8 nm. Below the distance of 4 nm between C60s,
graphene becomes locally suspended and less strained. We show that this happens when C60s
are allowed to deform under the compressive action of graphene. If we keep the C60s frozen,
spontaneous “blanketing” of graphene happens only when the distance between them are
equal or above 7 nm. Both above results for the existence of stable sagged graphene for C60
distances of 4 or 7 nm are shown to agree with a mechanical model relating the rigidity of
graphene to the energy of graphene-substrate adhesion. Although the studies of intercalation
of molecules on interfaces formed by graphene-substrate are motivated by finding out ways to
control wrinkling and strain in graphene, our work reveals the shape and structure of
intercalated molecules and the role of stability and wrinkling on final structure of graphene.
In particular, this study might help the development of 2D confined nanoreactors that are
considered in literature to be the next advanced step on chemical reactions.
AF; Dantas Fonseca, SO; Galvao
The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review) Journal Article
In: 2019.
BibTeX | Tags: C60, Graphene, Molecular Dynamics
@article{Fonseca2019c,
title = {The Structure of Graphene on Graphene/C60/Cu Interfaces: A Molecular Dynamics Study (under review)},
author = {Fonseca, AF; Dantas, SO; Galvao, DS; Zhang, D; Sinnott SB},
year = {2019},
date = {2019-03-15},
keywords = {C60, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Jaques, Ygor M.; Galvao, Douglas S.
Structural Properties of Nanodroplets Impacting Graphene at High Velocities (accepted) Journal Article
In: Journal of Molecular Liquids, 2019.
Abstract | BibTeX | Tags: droplets, Graphene, Impact Molecular Dynamics, water
@article{Jaques2019b,
title = {Structural Properties of Nanodroplets Impacting Graphene at High Velocities (accepted)},
author = {Ygor M. Jaques and Douglas S. Galvao},
year = {2019},
date = {2019-02-05},
journal = {Journal of Molecular Liquids},
abstract = {The determination of the wettability of 2D materials is an area of intensive research, as it is decisive on the applications of these systems in nanofluidics. One important part of the wetting characterization is how the spreading of droplets impacting on the surfaces occurs. However, few works address this problem for layered materials. Here, we report a fully atomistic molecular dynamics study on the dynamics of impact of water nanodroplets (100 ̊A of diameter) at high velocities (from 1 up to 15 ̊A/ps) against graphene targets. Our results show that tuning graphene wettability (through parameter changes) significantly affects the structural and dynamical aspects of the nanodroplets. We identified three ranges of velocities with distinct characteristics, from simple deposition of the droplet to spreading with rebound, and finally droplet frag- mentation. We also identify that in an intermediary velocity of 7 ̊A/ps, the pattern of spreading critically changes, due to formation of voids on droplet structure. These voids affect in a detrimental way the droplet spreading on the less hydrophilic surface, as it takes more time to the droplet recover from the spreading and to return to a semi-spherical configuration. When the velocity is increased to values larger than 11 ̊A/ps, the droplet fragments, which reveals the maximum possible spreading.},
keywords = {droplets, Graphene, Impact Molecular Dynamics, water},
pubstate = {published},
tppubtype = {article}
}

Fonseca, Alexandre F.; Galvao, Douglas S.
Self-tearing and self-peeling of folded graphene nanoribbons Journal Article
In: Carbon, vol. 143, pp. 230-239, 2019.
Abstract | Links | BibTeX | Tags: Fracture, Graphene, Mechanical Properties, Molecular Dynamics
@article{Fonseca2019,
title = {Self-tearing and self-peeling of folded graphene nanoribbons},
author = {Alexandre F. Fonseca and Douglas S. Galvao},
url = {https://www.sciencedirect.com/science/article/pii/S0008622318310431},
doi = {10.1016/j.carbon.2018.11.020},
year = {2019},
date = {2019-01-05},
journal = {Carbon},
volume = {143},
pages = {230-239},
abstract = {A recent experimental study showed that an induced folded flap of graphene can spontaneously drive itself its tearing and peeling off a substrate, thus producing long, micrometer sized, regular trapezoidal-shaped folded graphene nanoribbons. As long as the size of the graphene flaps is above a threshold value, the “tug of war” between the forces of adhesion of graphene-graphene and graphene-substrate, flexural strain of folded region and carbon-carbon (C-C) covalent bonds favor the self-tearing and self-peeling off process. As the detailed information regarding the atomic scale mechanism involved in the process remains not fully understood, we carried out atomistic reactive molecular dynamics simulations to address some features of the process. We show that large thermal fluctuations can prevent the process by increasing the probability of chemical reactions between carbon dangling bonds of adjacent graphene layers. The effects of the strength of attraction between graphene and the substrate on the ribbon growth velocities at the early stages of the phenomenon were also investigated. Structures with initial armchair crack-edges were observed to form more uniform cuts than those having initial zigzag ones. Our results are of importance to help set up new experiments on this phenomenon, especially with samples with nanoscale sized cuts. },
keywords = {Fracture, Graphene, Mechanical Properties, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
2018
Bizao, Rafael A; Machado, Leonardo D; de Sousa, Jose M; Pugno, Nicola M; Galvao, Douglas S
Scale Effects on the Ballistic Penetration of Graphene Sheets Journal Article
In: Nature Scientific Reports, vol. 8, pp. 6750, 2018.
Abstract | Links | BibTeX | Tags: Fracture, Graphene, impact, Molecular Dynamics
@article{Bizao2018,
title = {Scale Effects on the Ballistic Penetration of Graphene Sheets},
author = {Bizao, Rafael A and Machado, Leonardo D and de Sousa, Jose M and Pugno, Nicola M and Galvao, Douglas S},
url = {https://www.nature.com/articles/s41598-018-25050-2},
doi = {doi:10.1038/s41598-018-25050-2},
year = {2018},
date = {2018-04-30},
journal = {Nature Scientific Reports},
volume = {8},
pages = {6750},
abstract = {Carbon nanostructures are promising ballistic protection materials, due to their low density and excellent mechanical properties. Recent experimental and computational investigations on the behavior of graphene under impact conditions revealed exceptional energy absorption properties as well. However, the reported numerical and experimental values differ by an order of magnitude. In this work, we combined numerical and analytical modeling to address this issue. In the numerical part, we employed reactive molecular dynamics to carry out ballistic tests on single, double, and triple-layered graphene sheets. We used velocity values within the range tested in experiments. Our numerical and the experimental results were used to determine parameters for a scaling law. We find that the specific penetration energy decreases as the number of layers (N) increases, from ∼15 MJ/kg for N = 1 to ∼0.9 MJ/kg for N = 350, for an impact velocity of 900 m/s. These values are in good agreement with simulations and experiments, within the entire range of N values for which data is presently available. Scale effects explain the apparent discrepancy between simulations and experiments.},
keywords = {Fracture, Graphene, impact, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}

Ygor M.; Galvao Jaques, Douglas S.
Structural Properties of Nanodroplets Impacting Graphene at High Velocities Online
2018, (Preprint ArXiv:1804.07784).
Abstract | Links | BibTeX | Tags: droplets, Graphene, Impact Molecular Dynamics, water
@online{Jaques2018d,
title = {Structural Properties of Nanodroplets Impacting Graphene at High Velocities},
author = {Jaques, Ygor M.; Galvao, Douglas S.},
url = {https://arxiv.org/abs/1804.07784},
year = {2018},
date = {2018-04-24},
abstract = {We report here a fully atomistic molecular dynamics study on the dynamics of impact of water
nanodroplets (50, 100 and 120 Å of diameter) at high velocity (from 100 up to 1000 m/s) against
graphene targets. Our results show that tuning graphene wettability (through parameter changes)
significantly affects the structural and dynamical aspects of the nanodroplets. We identified three
ranges of velocities with distinct characteristics, from simple deposition of the droplet to
spreading with rebound and finally fragmentation. At Weber numbers lower than 10, the droplets
maintain a steady spreading factor independent of size. After this threshold value, the spread
rapidly grows with increasing Weber numbers. A more hydrophilic graphene surface increases
the spreading values, due to stronger solid-liquid interactions. Nevertheless, droplet size also
influences the fragmentation threshold, as an increased number of molecules make it easier for
the whole droplet overcomes the surface repulsion. },
note = {Preprint ArXiv:1804.07784},
keywords = {droplets, Graphene, Impact Molecular Dynamics, water},
pubstate = {published},
tppubtype = {online}
}
nanodroplets (50, 100 and 120 Å of diameter) at high velocity (from 100 up to 1000 m/s) against
graphene targets. Our results show that tuning graphene wettability (through parameter changes)
significantly affects the structural and dynamical aspects of the nanodroplets. We identified three
ranges of velocities with distinct characteristics, from simple deposition of the droplet to
spreading with rebound and finally fragmentation. At Weber numbers lower than 10, the droplets
maintain a steady spreading factor independent of size. After this threshold value, the spread
rapidly grows with increasing Weber numbers. A more hydrophilic graphene surface increases
the spreading values, due to stronger solid-liquid interactions. Nevertheless, droplet size also
influences the fragmentation threshold, as an increased number of molecules make it easier for
the whole droplet overcomes the surface repulsion.
Devi, M. Manolata; Dolai, N.; S, S. Sreehala; Jaques, Y. M.; Galvao, Douglas S.; C.S.Tiwary,; Sharma, Sudhanshu; Biswas, Krishanu
Morphology Controlled Graphene-Alloy Nanoparticles Hybrids with Tunable Catalytic Activity Journal Article
In: Nanoscale, vol. 10, pp. 8840-8850, 2018.
Abstract | Links | BibTeX | Tags: alloys, Graphene, Modeling, Nanoparticles
@article{Devi2018b,
title = {Morphology Controlled Graphene-Alloy Nanoparticles Hybrids with Tunable Catalytic Activity},
author = {M. Manolata Devi and N. Dolai and S. Sreehala S and Y. M. Jaques and Douglas S. Galvao and C.S.Tiwary and Sudhanshu Sharma and Krishanu Biswas},
url = {pubs.rsc.org/en/content/articlehtml/2018/nr/c7nr09688g},
doi = {10.1039/C7NR09688G},
year = {2018},
date = {2018-04-07},
journal = {Nanoscale},
volume = {10},
pages = {8840-8850},
abstract = {Selective oxidation of CO to CO2 using metallic or alloy nanoparticles as catalysts can solve two major problems of energy requirements and environmental pollution. Achieving 100% conversion efficiency at a lower temperature is a very important goal. This requires sustained efforts to design and develop novel supported catalysts containing alloy nanoparticles. In this regard, the decoration of nanoalloys with graphene, as a support for the catalyst, can provide a novel structure due to the synergic effect of the nanoalloys and graphene. Here, we demonstrate the effect of nano-PdPt (Palladium–Platinum) alloys having different morphologies on the catalytic efficiency for the selective oxidation of CO. Efforts were made to prepare different morphologies of PdPt alloy nanoparticles with the advantage of tuning the capping agent (PVP – polyvinyl pyrollidone) and decorating them on graphene sheets via the wet-chemical route. The catalytic activity of the G-PdPt hybrids with an urchin-like morphology has been found to be superior (higher % conversion at 135 °C lower) to that with a nanoflower morphology. The above experimental observations are further supported by molecular dynamics (MD) simulations.},
keywords = {alloys, Graphene, Modeling, Nanoparticles},
pubstate = {published},
tppubtype = {article}
}
Kabbani, Mohamad A.; Kochat, Vidya; Bhowmick, Sanjit; Soto, Matias; Som, Anirban; Krishnadas, K. R.; Woellner, Cristiano F.; Jaques, Ygor M.; Barrera, Enrique V.; Asif, Syed; Vajtai, Robert; Pradeep, Thalappil; Galvão, Douglas S.; Kabbani, Ahmad T.; Tiwary, Chandra Sekhar; Ajayan, Pulickel M.
Consolidation of Functionalized Graphene at Ambient Temperature via Mechano-chemistry Journal Article
In: Carbon, vol. 134, no. 8, pp. 491-499, 2018.
Abstract | Links | BibTeX | Tags: DFT, Graphene, Mechanochemistry, Molecular Dynamics
@article{Kabbani2018,
title = {Consolidation of Functionalized Graphene at Ambient Temperature via Mechano-chemistry},
author = {Mohamad A. Kabbani and Vidya Kochat and Sanjit Bhowmick and Matias Soto and Anirban Som and K.R. Krishnadas and Cristiano F. Woellner and Ygor M. Jaques and Enrique V. Barrera and Syed Asif and Robert Vajtai and Thalappil Pradeep and Douglas S. Galvão and Ahmad T. Kabbani and Chandra Sekhar Tiwary and Pulickel M. Ajayan},
url = {https://www.sciencedirect.com/science/article/pii/S0008622318302987?dgcid=raven_sd_aip_email},
doi = {DOI:10.1016/j.carbon.2018.03.049},
year = {2018},
date = {2018-03-22},
journal = {Carbon},
volume = {134},
number = {8},
pages = {491-499},
abstract = {Graphitic solids are typically produced via high temperature and energy consuming
processing (e.g. sintering) of carbon particles. Here, we demonstrate the mechano-chemical
assembly of functionalized graphene layers into 3D graphitic solids via room temperature and
low energy consuming processing. The chemical functional groups on graphene layers are
interconnected at room temperature under pressure leading to porous three-dimensional
structures with tunable mechanical and electrical properties. The formation of mechanochemistry
induced atomic scale junctions and their impact on mechanical properties of
graphene assembled carbon materials are demonstrated through nano-indentation experiments
and confirmed using DFT and molecular dynamics simulations. The results show room
temperature consolidation routes of graphene layers into bulk carbon solids.},
keywords = {DFT, Graphene, Mechanochemistry, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
processing (e.g. sintering) of carbon particles. Here, we demonstrate the mechano-chemical
assembly of functionalized graphene layers into 3D graphitic solids via room temperature and
low energy consuming processing. The chemical functional groups on graphene layers are
interconnected at room temperature under pressure leading to porous three-dimensional
structures with tunable mechanical and electrical properties. The formation of mechanochemistry
induced atomic scale junctions and their impact on mechanical properties of
graphene assembled carbon materials are demonstrated through nano-indentation experiments
and confirmed using DFT and molecular dynamics simulations. The results show room
temperature consolidation routes of graphene layers into bulk carbon solids.
Borges, Daiane Damasceno; Woellner, Cristiano F.; Autreto, Pedro A. S.; Galvao, Douglas S.
Water/alcohol separation via layered oxide graphene membranes Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 109-114, 2018.
Abstract | Links | BibTeX | Tags: Filtration, Graphene, Molecular Dynamics
@article{Borges2018d,
title = {Water/alcohol separation via layered oxide graphene membranes},
author = {Daiane Damasceno Borges and Cristiano F. Woellner and Pedro A. S. Autreto and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/wateralcohol-separation-in-graphene-oxide-membranes-insights-from-molecular-dynamics-and-monte-carlo-simulations/C61C66FF48D35EB2DB3408ACCE96C41A},
doi = { https://doi.org/10.1557/adv.2018.192},
year = {2018},
date = {2018-02-13},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = {109-114},
abstract = {Graphene-based membranes have been investigated as promising candidates for water filtration and gas separation applications. Experimental evidences have shown that graphene oxide can be impermeable to liquids, vapors and gases, while allowing a fast permeation of water molecules. This phenomenon has been attributed to the formation of a network of nano capillaries that allow nearly frictionless water flow while blocking other molecules by steric hindrance effects. It is supposed that water molecules are transported through the percolated two-dimensional channels formed between graphene-based sheets. Although these channels allow fast water permeation in such materials, the flow rates are strongly dependent on how the membranes are fabricated. Also, some fundamental issues regarding the nanoscale mechanisms of water permeation are still not fully understood and their interpretation remains controversial. In this work, we have investigated the dynamics of water permeation through pristine graphene and graphene oxide model membranes that have strong impact on water/alcohol separation. We have carried out fully atomistic classical molecular dynamics simulations of systems composed of multiple layered graphene-based sheets into contact with a pure water reservoir under controlled thermodynamics conditions (e. g., by varying temperature and pressure values). We have systematically analysed how the transport dynamics of the confined nanofluids depend on the interlayer distances and the role of the oxide functional groups. Our results show the water flux is much more effective for graphene than for graphene oxide membranes. These results can be attributed to the H-bonds formation between oxide functional groups and water, which traps the water molecules and precludes ultrafast water transport through the nanochannels.},
keywords = {Filtration, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Cristiano F Woellner Daiane Damasceno Borges, Pedro AS Autreto
Insights on the mechanism of water-alcohol separation in multilayer graphene oxide membranes: entropic versus enthalpic factors Journal Article
In: Carbon, vol. 127, pp. 280-286, 2018.
Abstract | Links | BibTeX | Tags: Filtration, Graphene, Molecular Dynamics
@article{Borges2018,
title = {Insights on the mechanism of water-alcohol separation in multilayer graphene oxide membranes: entropic versus enthalpic factors},
author = {Daiane Damasceno Borges, Cristiano F Woellner, Pedro AS Autreto, Douglas S Galvao},
url = {https://www.sciencedirect.com/science/article/pii/S000862231731134X},
doi = {https://doi.org/10.1016/j.carbon.2017.11.020},
year = {2018},
date = {2018-02-01},
journal = {Carbon},
volume = {127},
pages = {280-286},
abstract = {xperimental evidence has shown that graphene oxide (GO) can be impermeable to liquids, vapors and gases, while it allows a fast permeation of water molecules. Theoretical studies to understand the filtration mechanisms come mostly from water desalination, while very few works have been dedicated to alcohol dehydration. In this work, we have investigated the molecular level mechanism underlying the alcohol/water separation inside GO membranes. A series of Molecular Dynamics and Grand-Canonical Monte Carlo simulations were carried out to probe the ethanol/water and methanol/water separation through GO membranes composed of multiple layered graphene-based films with different interlayer distance values and number of oxygen-containing functional groups. Our results show that the size exclusion and membrane affinities are not sufficient to explain the selectivity. Besides that, the favorable water molecular arrangement inside GO 2D-channels forming a robust H-bond network and the fast water permeation are crucial for an effective separation mechanism. In other words, the separation phenomenon is not only governed by membrane affinities (enthalpic mechanisms) but mainly by the geometry and size factors (entropic mechanisms). Our findings are consistent with the available experimental data and contribute to clarify important aspects of the separation behavior of confined alcohol/water in GO membranes.},
keywords = {Filtration, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Fonseca, Alexandre F.; Galvao, Douglas S.
Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation Journal Article
In: MRS Advances, vol. 3, no. 8-9, pp. 460-465, 2018.
Abstract | Links | BibTeX | Tags: Fracture, Graphene, Molecular Dynamics
@article{Fonseca2018,
title = {Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation},
author = {Alexandre F. Fonseca and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/selfdriven-graphene-tearing-and-peeling-a-fully-atomistic-molecular-dynamics-investigation/BFC76FC4479AA617E16FA6AC7AB4D487},
doi = {https://doi.org/10.1557/adv.2018.120},
year = {2018},
date = {2018-01-30},
journal = {MRS Advances},
volume = {3},
number = {8-9},
pages = {460-465},
abstract = {In spite of years of intense research, graphene continues to produce surprising results. Recently, it was experimentally observed that under certain conditions graphene can self-drive its tearing and peeling from substrates. This process can generate long, micrometer sized, folded nanoribbons without the action of any external forces. Also, during this cracking-like propagation process, the width of the graphene folded ribbon continuously decreases and the process only stops when the width reaches about few hundreds nanometers in size. It is believed that interplay between the strain energy of folded regions, breaking of carbon-carbon covalent bonds, and adhesion of graphene-graphene and graphene-substrate are the most fundamental features of this process, although the detailed mechanisms at atomic scale remain unclear. In order to gain further insights on these processes we carried out fully atomistic reactive molecular dynamics simulations using the AIREBO potential as available in the LAMMPS computational package. Although the reported tearing/peeling experimental observations were only to micrometer sized structures, our results showed that they could also occur at nanometer scale. Our preliminary results suggest that the graphene tearing/peeling process originates from thermal energy fluctuations that results in broken bonds, followed by strain release that creates a local elastic wave that can either reinforce the process, similar to a whip cracking propagation, or undermine it by producing carbon dangling bonds that evolve to the formation of bonds between the two layers of graphene. As the process continues in time and the folded graphene decreases in width, the carbon-carbon bonds at the ribbon edge and interlayer bonds get less stressed, thermal fluctuations become unable to break them and the process stops.},
keywords = {Fracture, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Fonseca, Alexandre F.; Galvao, Douglas S.
Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation Online
2018, (preprint arXiv:1801.05354).
Abstract | Links | BibTeX | Tags: Fracture, Graphene, Molecular Dynamics
@online{Fonseca2018b,
title = {Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation},
author = {Alexandre F. Fonseca and Douglas S. Galvao
},
url = {https://arxiv.org/abs/1801.05354},
year = {2018},
date = {2018-01-17},
abstract = {In spite of years of intense research, graphene continues to produce surprising results. Recently, it was experimentally observed that under certain conditions graphene can self-drive its tearing and peeling from substrates. This process can generate long, micrometer sized, folded nanoribbons without the action of any external forces. Also, during this cracking-like propagation process, the width of the graphene folded ribbon continuously decreases and the process only stops when the width reaches about few hundreds nanometers in size. It is believed that interplay between the strain energy of folded regions, breaking of carbon-carbon covalent bonds, and adhesion of graphene-graphene and graphene-substrate are the most fundamental features of this process, although the detailed mechanisms at atomic scale remain unclear. In order to gain further insights on these processes we carried out fully atomistic reactive molecular dynamics simulations using the AIREBO potential as available in the LAMMPS computational package. Although the reported tearing/peeling experimental observations were only to micrometer sized structures, our results showed that they could also occur at nanometer scale. Our preliminary results suggest that the graphene tearing/peeling process originates from thermal energy fluctuations that results in broken bonds, followed by strain release that creates a local elastic wave that can either reinforce the process, similar to a whip cracking propagation, or undermine it by producing carbon dangling bonds that evolve to the formation of bonds between the two layers of graphene. As the process continues in time and the folded graphene decreases in width, the carbon-carbon bonds at the ribbon edge and interlayer bonds get less stressed, thermal fluctuations become unable to break them and the process stops.},
note = {preprint arXiv:1801.05354},
keywords = {Fracture, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}
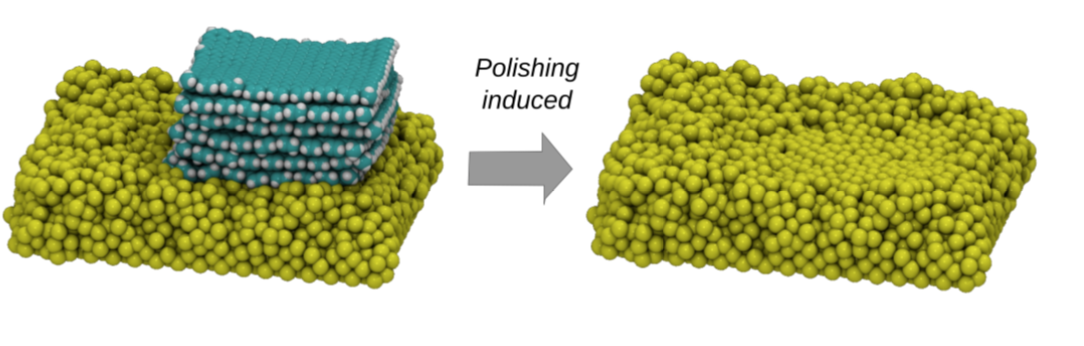
Oliveira, Eliezer Fernando; Santos, Ricardo Paupitz; da Silva Autreto, Pedro Alves; Stanislav Moshkalev,; Galvao, Douglas Soares
Improving Graphene-metal Contacts: Thermal Induced Polishing Online
2018, (preprint ArXiv:1801.04785).
Abstract | Links | BibTeX | Tags: contacts, Graphene, Molecular Dynamics, thermal properties
@online{Oliveira2018d,
title = {Improving Graphene-metal Contacts: Thermal Induced Polishing},
author = {Eliezer Fernando Oliveira and Ricardo Paupitz Santos and Pedro Alves da Silva Autreto and Stanislav Moshkalev, and Douglas Soares Galvao},
url = {https://arxiv.org/abs/1801.04785},
year = {2018},
date = {2018-01-15},
abstract = {Graphene is a very promising material for nanoelectronics applications due to its unique and remarkable electronic and thermal properties. However, when deposited on metallic electrodes the overall thermal conductivity is significantly decreased. This phenomenon has been attributed to the mismatch between the interfaces and contact thermal resistance. Experimentally, one way to improve the graphene/metal contact is thorough high-temperature annealing, but the detailed mechanisms behind these processes remain unclear. In order to address these questions, we carried out fully atomistic reactive molecular dynamics simulations using the ReaxFF force field to investigate the interactions between multi-layer graphene and metallic electrodes (nickel) under (thermal) annealing. Our results show that the annealing induces an upward-downward movement of the graphene layers, causing a pile- driver-like effect over the metallic surface. This graphene induced movements cause a planarization (thermal polishing-like effect) of the metallic surface, which results in the increase of the effective graphene/metal contact area. This can also explain the experimentally observed improvements of the thermal and electric conductivities.},
note = {preprint ArXiv:1801.04785},
keywords = {contacts, Graphene, Molecular Dynamics, thermal properties},
pubstate = {published},
tppubtype = {online}
}
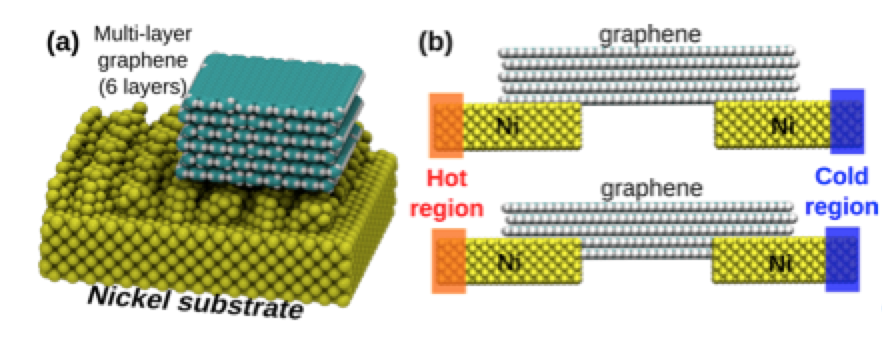
Oliveira, Eliezer Fernando; Paupitz, Ricardo; da Silva Autreto, Pedro Alves; Moshkalev, Stanislav; Galvao, Douglas Soares
Improving Graphene-metal Contacts: Thermal Induced Polishing Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 73-78, 2018.
Abstract | Links | BibTeX | Tags: contacts, Graphene, Molecular Dynamics, thermal properties
@article{Oliveira2018c,
title = {Improving Graphene-metal Contacts: Thermal Induced Polishing },
author = {Eliezer Fernando Oliveira and Ricardo Paupitz and Pedro Alves da Silva Autreto and Stanislav Moshkalev and Douglas Soares Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/improving-graphenemetal-contacts-thermal-induced-polishing/AC01C4996B90B0EE5E03220604071D12},
doi = {https://doi.org/10.1557/adv.2018.66},
year = {2018},
date = {2018-01-01},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = {73-78},
abstract = {Graphene is a very promising material for nanoelectronics applications due to its unique and remarkable electronic and thermal properties. However, when deposited on metallic electrodes the overall thermal conductivity is significantly decreased. This phenomenon has been attributed to the mismatch between the interfaces and contact thermal resistance. Experimentally, one way to improve the graphene/metal contact is thorough high-temperature annealing, but the detailed mechanisms behind these processes remain unclear. In order to address these questions, we carried out fully atomistic reactive molecular dynamics simulations using the ReaxFF force field to investigate the interactions between multi-layer graphene and metallic electrodes (nickel) under (thermal) annealing. Our results show that the annealing induces an upward-downward movement of the graphene layers, causing a pile-driver-like effect over the metallic surface. This graphene induced movements cause a planarization (thermal polishing-like effect) of the metallic surface, which results in the increase of the effective graphene/metal contact area. This can also explain the experimentally observed improvements of the thermal and electric conductivities.},
keywords = {contacts, Graphene, Molecular Dynamics, thermal properties},
pubstate = {published},
tppubtype = {article}
}
2017
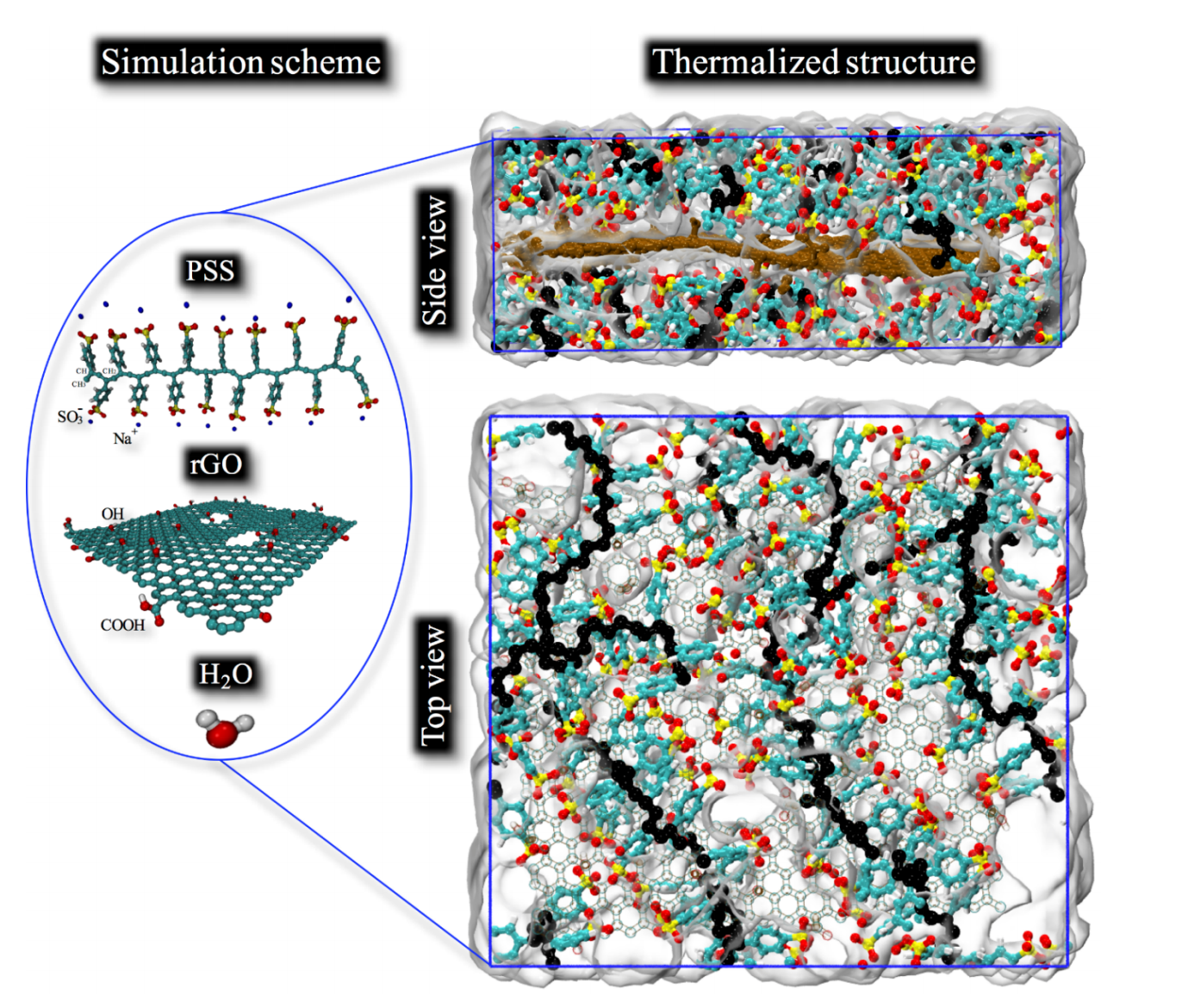
Miyazaki, Celina M; Maria, Marco AE; Borges, Daiane Damasceno; Woellner, Cristiano F; Brunetto, Gustavo; Fonseca, Alexandre F; Constantino, Carlos JL; Pereira-da-Silva, Marcelo A; de Siervo, Abner; Galvao, Douglas S; Riul Jr., Antonio
2017, (preprint arXiv:1702.00250).
Abstract | Links | BibTeX | Tags: Graphene, Molecular Dynamics, Polymers
@online{Miyazaki2017,
title = {Synthesis, characterization and computational simulation of graphene nanoplatelets stabilized in poly (styrene sulfonate) sodium salt},
author = {Miyazaki, Celina M and Maria, Marco AE and Borges, Daiane Damasceno and Woellner, Cristiano F and Brunetto, Gustavo and Fonseca, Alexandre F and Constantino, Carlos JL and Pereira-da-Silva, Marcelo A and de Siervo, Abner and Galvao, Douglas S and Riul Jr., Antonio},
url = {https://arxiv.org/abs/1705.10673},
year = {2017},
date = {2017-05-30},
abstract = {The production of large area interfaces and the use of scalable methods to build-up designed nanostructures generating advanced functional properties are of high interest for many materials science applications. Nevertheless, large area coverage remains a major problem for pristine graphene and here we present a hybrid, composite graphene-like material soluble in water, which can be exploited in many areas, such as energy storage, electrodes fabrication, selective membranes and biosensing. Graphene oxide (GO) was produced by the traditional Hummers method being further reduced in the presence of poly(styrene sulfonate) sodium salt (PSS), thus creating stable reduced graphene oxide (rGO) nanoplateles wrapped by PSS (GPSS). Molecular dynamics simulations were carried out of further clarify the interactions between PSS molecules and rGO nanoplatelets, with calculations supported by FTIR analysis. The intermolecular forces between rGO nanoplatelets and PSS lead to the formation of a hybrid material (GPSS) stabilized by van der Waals forces, allowing the fabrication of high quality layer-by-layer (LbL) films with polyalillamine hydrochloride (PAH). Raman and electrical characterizations corroborated the successful modifications in the electronic structures from GO to GPSS after the chemical treatment, resulting in (PAH/GPSS) LbL films four orders of magnitude more conductive than (PAH/GO).
},
note = {preprint arXiv:1702.00250},
keywords = {Graphene, Molecular Dynamics, Polymers},
pubstate = {published},
tppubtype = {online}
}
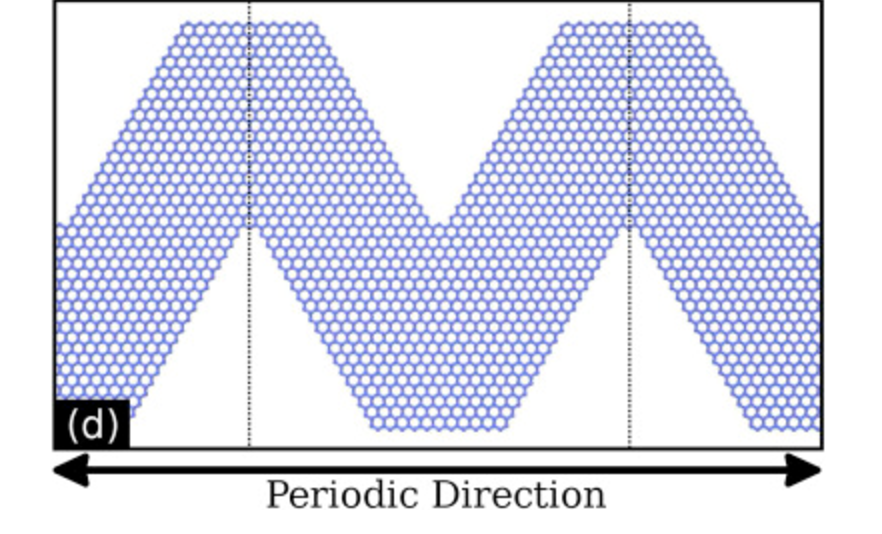
Bizao, Rafael A; Botari, Tiago; Perim, Eric; Pugno, Nicola M; Galvao, Douglas S
Mechanical properties and fracture patterns of graphene (graphitic) nanowiggles Journal Article
In: Carbon, vol. 119, pp. 431-437, 2017, (See also ArxIv version: https://arxiv.org/abs/1702.01100).
Abstract | Links | BibTeX | Tags: Graphene, Molecular Dynamics, NanoRibbons, Nanowiggles
@article{Bizao2017b,
title = {Mechanical properties and fracture patterns of graphene (graphitic) nanowiggles},
author = {Bizao, Rafael A and Botari, Tiago and Perim, Eric and Pugno, Nicola M and Galvao, Douglas S},
url = {http://www.sciencedirect.com/science/article/pii/S0008622317303743},
doi = {10.1016/j.carbon.2017.04.018},
year = {2017},
date = {2017-04-14},
journal = {Carbon},
volume = {119},
pages = {431-437},
abstract = {Graphene nanowiggles (GNW) are graphene-based nanostructures obtained by making alternated regular cuts in pristine graphene nanoribbons. GNW were recently synthesized and it was demonstrated that they exhibit tunable electronic and magnetic properties by just varying their shape. Here, we have investigated the mechanical properties and fracture patterns of a large number of GNW of different shapes and sizes using fully atomistic reactive molecular dynamics simulations. Our results show that the GNW mechanical properties are strongly dependent on its shape and size and, as a general trend narrow sheets have larger ultimate strength and Young's modulus than wide ones. The estimated Young's modulus values were found to be in a range of ≈100−1000 GPa and the ultimate strength in a range of ≈20−110 GPa, depending on GNW shape. Also, super-ductile behavior under strain was observed for some structures.},
note = {See also ArxIv version: https://arxiv.org/abs/1702.01100},
keywords = {Graphene, Molecular Dynamics, NanoRibbons, Nanowiggles},
pubstate = {published},
tppubtype = {article}
}
Splugues, Vinicius; da Silva Autreto, Pedro Alves; Galvao, Douglas S
Hydrogenation Dynamics of Biphenylene Carbon (Graphenylene) Membranes Journal Article
In: MRS Advances, vol. 2017, pp. 1-6, 2017.
Abstract | Links | BibTeX | Tags: Graphene, Hydrogenation, Molecular Dynamics
@article{Splugues2017,
title = {Hydrogenation Dynamics of Biphenylene Carbon (Graphenylene) Membranes},
author = {Splugues, Vinicius and da Silva Autreto, Pedro Alves and Galvao, Douglas S},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/hydrogenation-dynamics-of-biphenylene-carbon-graphenylene-membranes/139DB900D41560D64F352A31CE219D3A},
doi = {10.1557/adv.2017.239},
year = {2017},
date = {2017-02-28},
journal = {MRS Advances},
volume = {2017},
pages = {1-6},
abstract = {The advent of graphene created a revolution in materials science. Because of this there is a renewed interest in other carbon-based structures. Graphene is the ultimate (just one atom thick) membrane. It has been proposed that graphene can work as impermeable membrane to standard gases, such argon and helium. Graphene-like porous membranes, but presenting larger porosity and potential selectivity would have many technological applications. Biphenylene carbon (BPC), sometimes called graphenylene, is one of these structures. BPC is a porous two-dimensional (planar) allotrope carbon, with its pores resembling typical sieve cavities and/or some kind of zeolites. In this work, we have investigated the hydrogenation dynamics of BPC membranes under different conditions (hydrogenation plasma density, temperature, etc.). We have carried out an extensive study through fully atomistic molecular dynamics (MD) simulations using the reactive force field ReaxFF, as implemented in the well-known Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) code. Our results show that the BPC hydrogenation processes exhibit very complex patterns and the formation of correlated domains (hydrogenated islands) observed in the case of graphene hydrogenation was also observed here. MD results also show that under hydrogenation BPC structure undergoes a change in its topology, the pores undergoing structural transformations and extensive hydrogenation can produce significant structural damages, with the formation of large defective areas and large structural holes, leading to structural collapse.
},
keywords = {Graphene, Hydrogenation, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
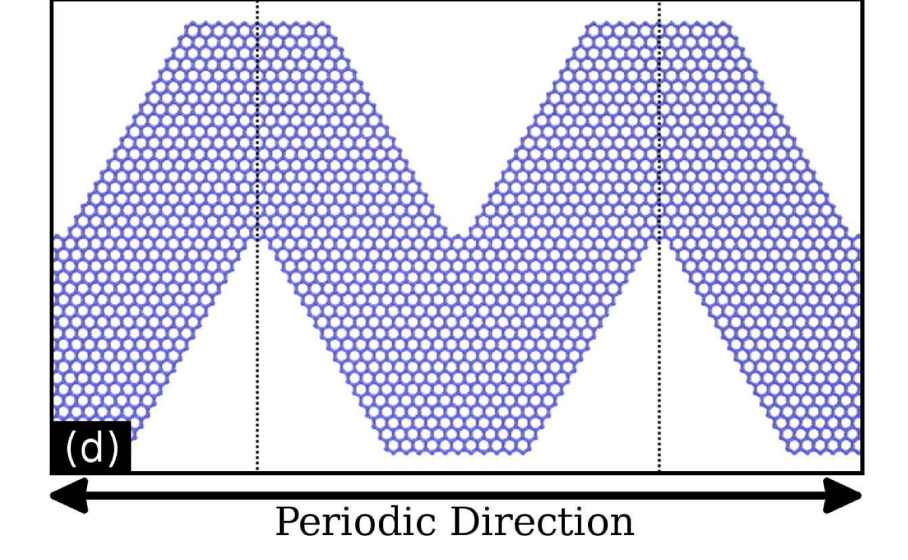
Bizao, Rafael A; Botari, Tiago; Perim, Eric; Pugno, Nicola M; Galvao, Douglas S
Mechanical Properties and Fracture Patterns of Graphene (Graphitic) Nanowiggles Online
2017, (preprint arXiv:1702.01100).
Abstract | Links | BibTeX | Tags: Graphene, Mechanical Properties, Molecular Dynamics, Nanowiggles
@online{Bizao2017,
title = {Mechanical Properties and Fracture Patterns of Graphene (Graphitic) Nanowiggles},
author = {Bizao, Rafael A and Botari, Tiago and Perim, Eric and Pugno, Nicola M and Galvao, Douglas S},
url = {https://arxiv.org/pdf/1702.01100.pdf},
year = {2017},
date = {2017-02-03},
abstract = {Graphene nanowiggles (GNW) are graphene-based nanostructures
obtained by making alternated regular cuts in pristine graphene nanoribbons.
GNW were recently synthesized and it was demonstrated that
they exhibit tunable electronic and magnetic properties by just varying
their shape. Here, we have investigated the mechanical properties and
fracture patterns of a large number of GNW of different shapes and
sizes using fully atomistic reactive molecular dynamics simulations.
Our results show that the GNW mechanical properties are strongly
dependent on its shape and size and, as a general trend narrow sheets
have larger ultimate strength and Young’s modulus than wide ones.
The estimated Young’s modulus values were found to be in a range of
≈ 100 − 1000 GPa and the ultimate strength in a range of ≈ 20 − 110
GPa, depending on GNW shape. Also, super-ductile behaviour under
strain was observed for some structures.},
note = {preprint arXiv:1702.01100},
keywords = {Graphene, Mechanical Properties, Molecular Dynamics, Nanowiggles},
pubstate = {published},
tppubtype = {online}
}
obtained by making alternated regular cuts in pristine graphene nanoribbons.
GNW were recently synthesized and it was demonstrated that
they exhibit tunable electronic and magnetic properties by just varying
their shape. Here, we have investigated the mechanical properties and
fracture patterns of a large number of GNW of different shapes and
sizes using fully atomistic reactive molecular dynamics simulations.
Our results show that the GNW mechanical properties are strongly
dependent on its shape and size and, as a general trend narrow sheets
have larger ultimate strength and Young’s modulus than wide ones.
The estimated Young’s modulus values were found to be in a range of
≈ 100 − 1000 GPa and the ultimate strength in a range of ≈ 20 − 110
GPa, depending on GNW shape. Also, super-ductile behaviour under
strain was observed for some structures.
Borges, Daiane D; Woellner, Cristiano F; Autreto, Pedro AS; Galvao, Douglas S
2017, (preprint arXiv:1702.00250).
Abstract | Links | BibTeX | Tags: Graphene, Molecular Dynamics, water
@online{Borges2017,
title = {Water Permeation through Layered Graphene-based Membranes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Borges, Daiane D and Woellner, Cristiano F and Autreto, Pedro AS and Galvao, Douglas S},
url = {https://arxiv.org/abs/1702.00250},
year = {2017},
date = {2017-02-01},
abstract = {Graphene-based membranes have been investigated as promising candidates for water
filtration and gas separation applications. Experimental evidences have shown that graphene
oxide can be impermeable to liquids, vapors and gases, while allowing a fast permeation of water
molecules. This phenomenon has been attributed to the formation of a network of nano
capillaries that allow nearly frictionless water flow while blocking other molecules by steric
hindrance effects. It is supposed that water molecules are transported through the percolated twodimensional
channels formed between graphene-based sheets. Although these channels allow
fast water permeation in such materials, the flow rates are strongly dependent on how the
membranes are fabricated. Also, some fundamental issues regarding the nanoscale mechanisms
of water permeation are still not fully understood and their interpretation remains controversial.
In this work, we have investigated the dynamics of water permeation through pristine graphene
and graphene oxide model membranes. We have carried out fully atomistic classical molecular
dynamics simulations of systems composed of multiple layered graphene-based sheets into
contact with a water reservoir under controlled thermodynamics conditions (e. g., by varying
temperature and pressure values). We have systematically analyzed how the transport dynamics
of the confined nanofluids depend on the interlayer distances and the role of the oxide functional
groups. Our results show the water flux is much more effective for graphene than for graphene
oxide membranes. These results are attributed to the H-bonds formation between oxide
functional groups and water, which traps the water molecules and precludes ultrafast water
transport through the nanochannels.},
note = {preprint arXiv:1702.00250},
keywords = {Graphene, Molecular Dynamics, water},
pubstate = {published},
tppubtype = {online}
}
filtration and gas separation applications. Experimental evidences have shown that graphene
oxide can be impermeable to liquids, vapors and gases, while allowing a fast permeation of water
molecules. This phenomenon has been attributed to the formation of a network of nano
capillaries that allow nearly frictionless water flow while blocking other molecules by steric
hindrance effects. It is supposed that water molecules are transported through the percolated twodimensional
channels formed between graphene-based sheets. Although these channels allow
fast water permeation in such materials, the flow rates are strongly dependent on how the
membranes are fabricated. Also, some fundamental issues regarding the nanoscale mechanisms
of water permeation are still not fully understood and their interpretation remains controversial.
In this work, we have investigated the dynamics of water permeation through pristine graphene
and graphene oxide model membranes. We have carried out fully atomistic classical molecular
dynamics simulations of systems composed of multiple layered graphene-based sheets into
contact with a water reservoir under controlled thermodynamics conditions (e. g., by varying
temperature and pressure values). We have systematically analyzed how the transport dynamics
of the confined nanofluids depend on the interlayer distances and the role of the oxide functional
groups. Our results show the water flux is much more effective for graphene than for graphene
oxide membranes. These results are attributed to the H-bonds formation between oxide
functional groups and water, which traps the water molecules and precludes ultrafast water
transport through the nanochannels.

Bizao, Rafael A; Machado, Leonardo D; de Sousa, Jose M; Pugno, Nicola M; Galvao, Douglas S
Scale Effects on the Ballistic Penetration of Graphene Sheets Online
2017, (preprint arXiv:1701.07367).
Abstract | Links | BibTeX | Tags: ballistic impacts, Fracture, Graphene, Molecular Dynamics
@online{Bizao2017c,
title = {Scale Effects on the Ballistic Penetration of Graphene Sheets},
author = {Bizao, Rafael A and Machado, Leonardo D and de Sousa, Jose M and Pugno, Nicola M and Galvao, Douglas S},
url = {https://arxiv.org/pdf/1701.07367.pdf},
year = {2017},
date = {2017-01-25},
abstract = {Carbon nanostructures are promising ballistic protection materials,
due to their low density and excellent mechanical properties. Recent
experimental and computational investigations on the behavior
of graphene under impact conditions revealed exceptional energy absorption
properties as well. However, the reported numerical and experimental
values differ by an order of magnitude. In this work, we
combined numerical and analytical modeling to address this issue. In
the numerical part, we employed reactive molecular dynamics to carry
out ballistic tests on single and double-layered graphene sheets. We
used velocity values within the range tested in experiments. Our numerical
and the experimental results were used to determine parameters
for a scaling law, which is in good agreement with all experimental
and simulation results. We find that the specific penetration energy
decreases as the number of layers (N) increases, from ∼ 25 MJ/kg for
N = 1 to ∼ 0.26 MJ/kg as N → ∞. These scale effects explain the
apparent discrepancy between simulations and experiments.},
note = {preprint arXiv:1701.07367},
keywords = {ballistic impacts, Fracture, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}
due to their low density and excellent mechanical properties. Recent
experimental and computational investigations on the behavior
of graphene under impact conditions revealed exceptional energy absorption
properties as well. However, the reported numerical and experimental
values differ by an order of magnitude. In this work, we
combined numerical and analytical modeling to address this issue. In
the numerical part, we employed reactive molecular dynamics to carry
out ballistic tests on single and double-layered graphene sheets. We
used velocity values within the range tested in experiments. Our numerical
and the experimental results were used to determine parameters
for a scaling law, which is in good agreement with all experimental
and simulation results. We find that the specific penetration energy
decreases as the number of layers (N) increases, from ∼ 25 MJ/kg for
N = 1 to ∼ 0.26 MJ/kg as N → ∞. These scale effects explain the
apparent discrepancy between simulations and experiments.
2016
Chandra Sekhar Tiwary Dibyendu Chakravarty, Cristano F Woellner
3D Porous Graphene by Low-Temperature Plasma Welding for Bone Implants Journal Article
In: Advanced Materials, vol. 28, no. 40, pp. 8959-8967, 2016.
Abstract | Links | BibTeX | Tags: Graphene, Molecular Dynamics, Plasma Welding
@article{chakravarty20163d,
title = {3D Porous Graphene by Low-Temperature Plasma Welding for Bone Implants},
author = {Dibyendu Chakravarty, Chandra Sekhar Tiwary, Cristano F Woellner, Sruthi Radhakrishnan, Soumya Vinod, Sehmus Ozden, Pedro Alves da Silva Autreto, Sanjit Bhowmick, Syed Asif, Sendurai A Mani, Douglas S Galvao, Pulickel M},
url = {onlinelibrary.wiley.com/doi/10.1002/adma.201603146/abstract },
doi = {10.1002/adma.201603146},
year = {2016},
date = {2016-08-26},
journal = {Advanced Materials},
volume = {28},
number = {40},
pages = {8959-8967},
abstract = {3D scaffolds of graphene, possessing ultra-low density, macroporous microstructure, and high yield strength and stiffness can be developed by a novel plasma welding process. The bonding between adjacent graphene sheets is investigated by molecular dynamics simulations. The high degree of biocompatibility along with high porosity and good mechanical properties makes graphene an ideal material for use as body implants.},
keywords = {Graphene, Molecular Dynamics, Plasma Welding},
pubstate = {published},
tppubtype = {article}
}
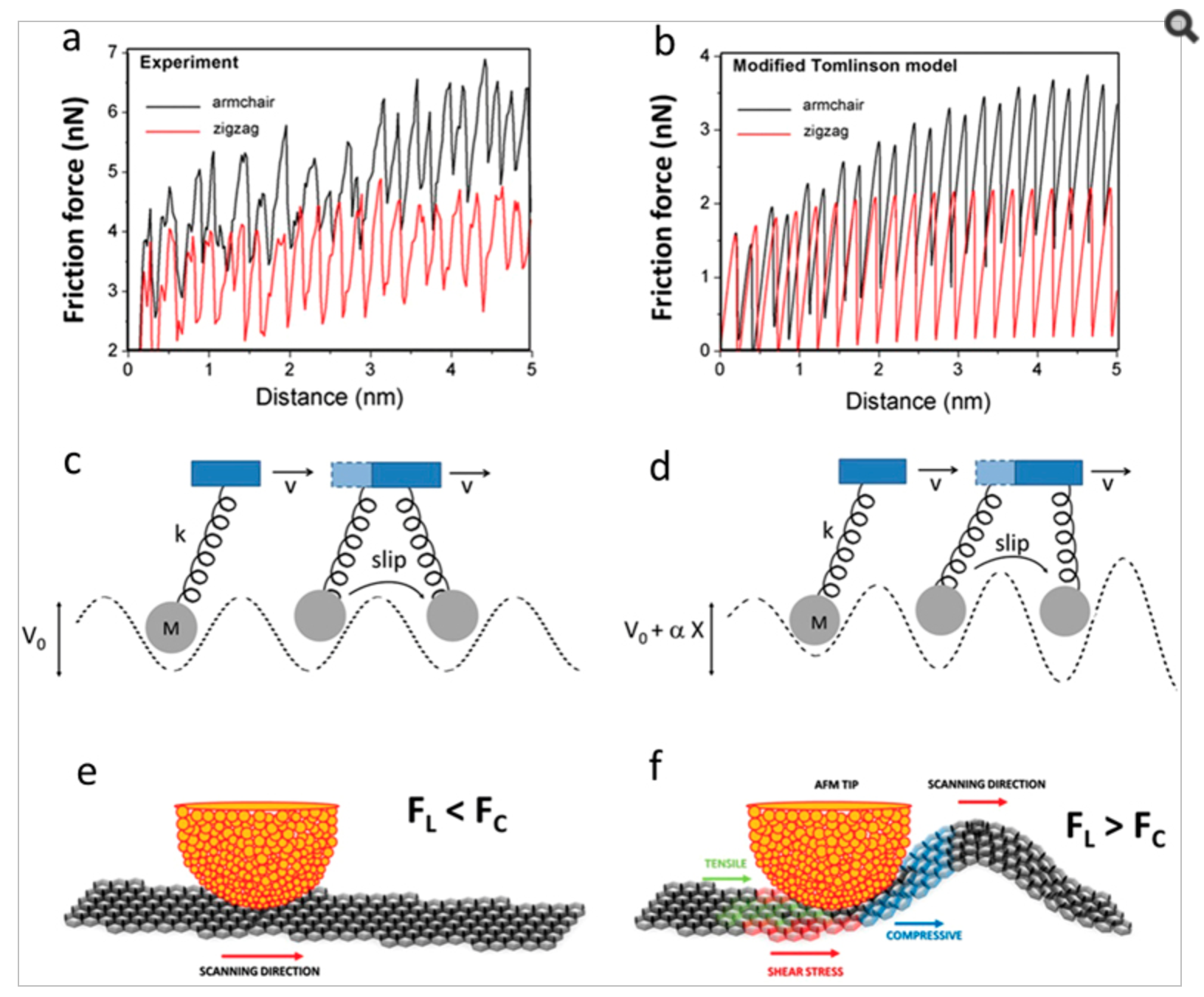
Rodrigo Prioli Clara M Almeida, Benjamin Fragneaud
Giant and Tunable Anisotropy of Nanoscale Friction in Graphene Journal Article
In: Nature Scientific Reports, vol. 6, pp. 31569, 2016.
Abstract | Links | BibTeX | Tags: DFT, Graphene, Molecular Dynamics, Tribology
@article{Almeida2016,
title = {Giant and Tunable Anisotropy of Nanoscale Friction in Graphene},
author = {Clara M Almeida, Rodrigo Prioli, Benjamin Fragneaud, Luiz Gustavo Cançado, Ricardo Paupitz, Douglas S Galvão, Marcelo De Cicco, Marcos G Menezes, Carlos A Achete, Rodrigo B Capaz},
url = {http://www-nature-com.ez88.periodicos.capes.gov.br/articles/srep31569},
doi = {10.1038/srep31569},
year = {2016},
date = {2016-07-18},
journal = {Nature Scientific Reports},
volume = {6},
pages = {31569},
abstract = {The nanoscale friction between an atomic force microscopy tip and graphene is investigated using friction force microscopy (FFM). During the tip movement, friction forces are observed to increase and then saturate in a highly anisotropic manner. As a result, the friction forces in graphene are highly dependent on the scanning direction: under some conditions, the energy dissipated along the armchair direction can be 80% higher than along the zigzag direction. In comparison, for highly-oriented pyrolitic graphite (HOPG), the friction anisotropy between armchair and zigzag directions is only 15%. This giant friction anisotropy in graphene results from anisotropies in the amplitudes of flexural deformations of the graphene sheet driven by the tip movement, not present in HOPG. The effect can be seen as a novel manifestation of the classical phenomenon of Euler buckling at the nanoscale, which provides the non-linear ingredients that amplify friction anisotropy. Simulations based on a novel version of the 2D Tomlinson model (modified to include the effects of flexural deformations), as well as fully atomistic molecular dynamics simulations and first-principles density-functional theory (DFT) calculations, are able to reproduce and explain the experimental observations.
},
keywords = {DFT, Graphene, Molecular Dynamics, Tribology},
pubstate = {published},
tppubtype = {article}
}
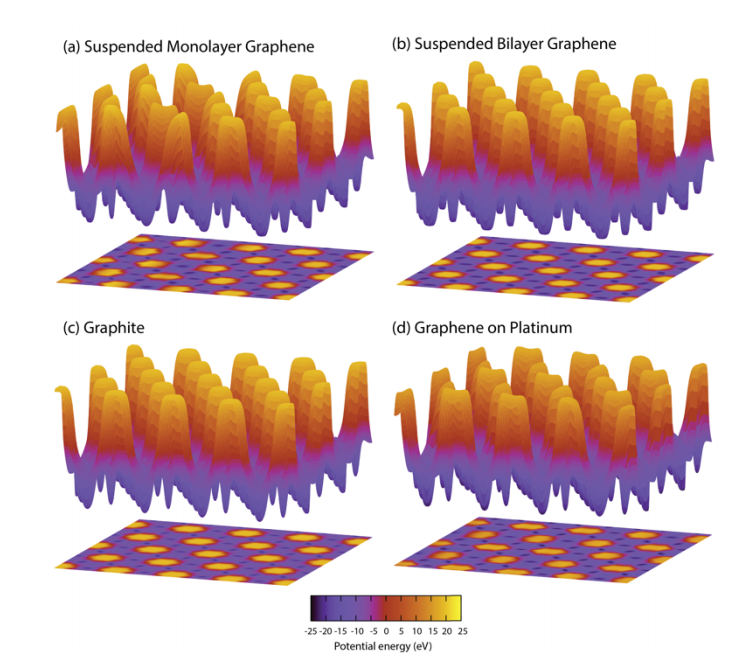
Pedro Alves da Silva Autreto Cristiano Francisco Woellner, Douglas S Galvao
Graphone (one-side hydrogenated graphene) formation on different substrates Online
2016.
Abstract | Links | BibTeX | Tags: Graphene, graphone, Molecular Dynamics
@online{Woellner2016b,
title = {Graphone (one-side hydrogenated graphene) formation on different substrates},
author = {Cristiano Francisco Woellner, Pedro Alves da Silva Autreto, Douglas S Galvao},
url = {arXiv preprint arXiv:1606.09235},
year = {2016},
date = {2016-06-29},
abstract = {In this work we present a fully atomistic reactive (ReaxFF force field) molecular dynamics study of the structural and dynamical aspects of the one-side hydrogenation of graphene membranes, leading to the formation of the so-called graphone structure. We have considered different substrates: graphene, few-layers graphene, graphite and platinum at different temperatures. Our results showed that the hydrogenation rates are very dependent on the substrate and thermal effects. Our results also showed that, similarly to graphane, large hydrogenated domains are unlikely to be formed. These hydrogenation processes occur through the formation of uncorrelated cluster domains.},
keywords = {Graphene, graphone, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}
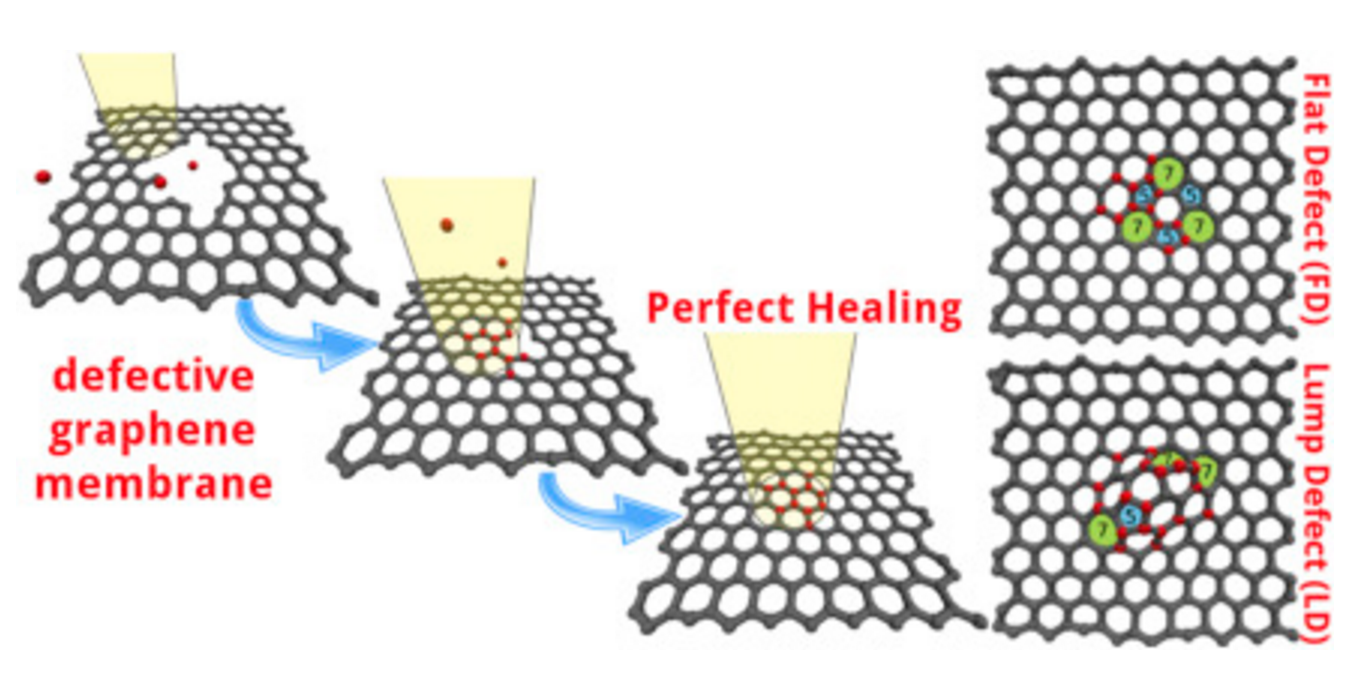
Botari, Tiago; Paupitz, Ricardo; da Silva Autreto, Pedro Alves; Galvao, Douglas S
Graphene healing mechanisms: A theoretical investigation Journal Article
In: Carbon, vol. 99, pp. 302-309, 2016.
Abstract | Links | BibTeX | Tags: Graphene, healing, Molecular Dynamics
@article{2016Healing,
title = {Graphene healing mechanisms: A theoretical investigation},
author = {Botari, Tiago and Paupitz, Ricardo and da Silva Autreto, Pedro Alves and Galvao, Douglas S},
url = {http://www.sciencedirect.com/science/article/pii/S0008622315304784},
doi = {10.1016/j.carbon.2015.11.070},
year = {2016},
date = {2016-04-30},
journal = {Carbon},
volume = {99},
pages = {302-309},
abstract = {Large holes in graphene membranes were recently shown to heal, either at room temperature during a low energy STEM experiment, or by annealing at high temperatures. However, the details of the healing mechanism remain unclear. We carried out fully atomistic reactive molecular dynamics simulations in order to address these mechanisms under different experimental conditions. Our results show that, if a carbon atom source is present, high temperatures can provide enough energy for the carbon atoms to overcome the potential energy barrier and to produce perfect reconstruction of the graphene hexagonal structure. At room temperature, this perfect healing is only possible if the heat effects of the electron beam from STEM experiment are explicitly taken into account. The reconstruction process of a perfect or near perfect graphene structure involves the formation of linear carbon chains, as well as rings containing 5, 6, 7 and 8 atoms with planar (Stone-Wales like) and non-planar (lump like) structures. These results shed light on the healing mechanism of graphene when subjected to different experimental conditions. Additionally, the methodology presented here can be useful for investigating the tailoring and manipulations of other nano-structures.},
keywords = {Graphene, healing, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
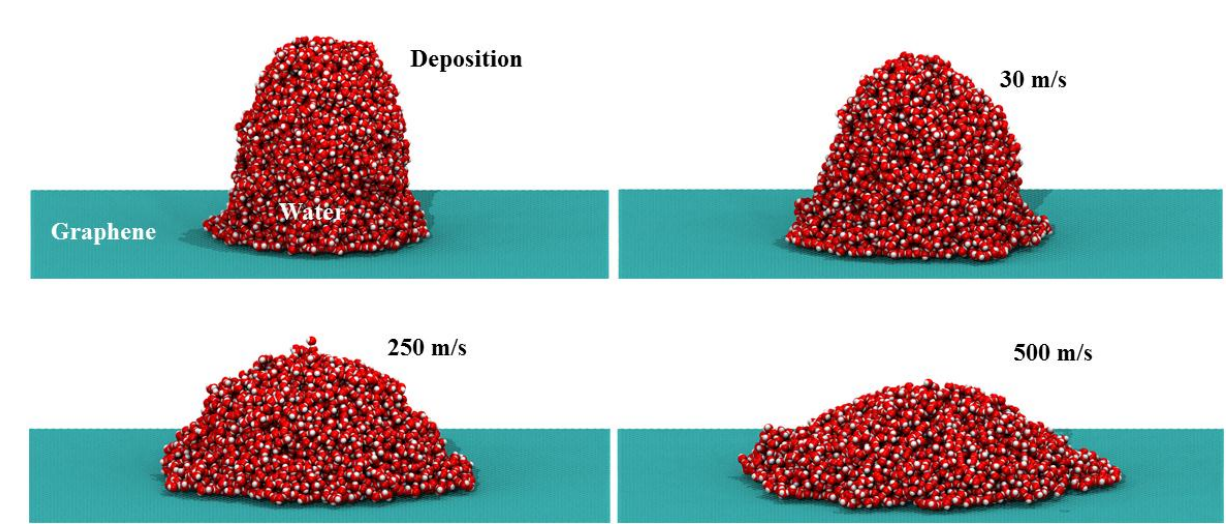
Ygor M. Jaques, Gustavo Brunetto; Galvão, Douglas S.
Nanodroplets Impacting on Graphene Journal Article
In: MRS Advances, vol. 2016, 2016.
Abstract | Links | BibTeX | Tags: Graphene, Impact Molecular Dynamics, nanodroplet
@article{Jaques2016b,
title = {Nanodroplets Impacting on Graphene},
author = {Ygor M. Jaques, Gustavo Brunetto and Douglas S. Galvão},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=10253580&fulltextType=RA&fileId=S2059852116002218},
doi = {DOI: 10.1557/adv.2016.221},
year = {2016},
date = {2016-03-01},
journal = {MRS Advances},
volume = {2016},
abstract = {The unique and remarkable properties of graphene can be exploited as the basis to a wide
range of applications. However, in spite of years of investigations there are some important
graphene properties that are not still fully understood, as for example, its wettability. There are
controversial reported results whether graphene is really hydrophobic or hydrophilic. In order to
address this problem we have carried out classical molecular dynamics simulations of water
nanodroplets shot against graphene surface. Our results show that the contact angle values
between the nanodroplets and graphene surfaces depend on the initial droplet velocity value and
these angles can change from 86º (hydrophobic) to 35º (hydrophilic). Our preliminary results
indicate that the graphene wettability can be dependent on spreading liquid dynamics and which
can explain some of the apparent inconsistencies reported in the literature.},
keywords = {Graphene, Impact Molecular Dynamics, nanodroplet},
pubstate = {published},
tppubtype = {article}
}
range of applications. However, in spite of years of investigations there are some important
graphene properties that are not still fully understood, as for example, its wettability. There are
controversial reported results whether graphene is really hydrophobic or hydrophilic. In order to
address this problem we have carried out classical molecular dynamics simulations of water
nanodroplets shot against graphene surface. Our results show that the contact angle values
between the nanodroplets and graphene surfaces depend on the initial droplet velocity value and
these angles can change from 86º (hydrophobic) to 35º (hydrophilic). Our preliminary results
indicate that the graphene wettability can be dependent on spreading liquid dynamics and which
can explain some of the apparent inconsistencies reported in the literature.
Pedro Alves da Silva Autreto Cristiano Francisco Woellner, Douglas S. Galvao
One Side-Graphene Hydrogenation (Graphone): Substrate Effects Journal Article
In: MRS Advances, vol. 2016, 2016.
Abstract | Links | BibTeX | Tags: Graphane, Graphene, graphone, Molecular Dynamics
@article{Woellner2016b,
title = {One Side-Graphene Hydrogenation (Graphone): Substrate Effects},
author = {Cristiano Francisco Woellner, Pedro Alves da Silva Autreto, Douglas S. Galvao},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=10234793&fulltextType=RA&fileId=S2059852116001961},
doi = {DOI: 10.1557/adv.2016.196},
year = {2016},
date = {2016-03-01},
journal = {MRS Advances},
volume = {2016},
abstract = {Recent studies on graphene hydrogenation processes showed that hydrogenation occurs via island growing domains, however how the substrate can affect the hydrogenation dynamics and/or pattern formation has not been yet properly investigated. In this work we have addressed these issues through fully atomistic reactive molecular dynamics simulations. We investigated the structural and dynamical aspects of the hydrogenation of graphene membranes (one-side hydrogenation, the so called graphone structure) on different substrates (graphene, few-layers graphene, graphite and platinum). Our results also show that the observed hydrogenation rates are very sensitive to the substrate type. For all investigated cases, the largest fraction of hydrogenated carbon atoms was for platinum substrates. Our results also show that a significant number of randomly distributed H clusters are formed during the early stages of the hydrogenation process, regardless of the type of substrate. These results suggest that, similarly to graphane formation, large perfect graphone-like domains are unlikely to be formed. These findings are especially important since experiments have showed that cluster formation influences the electronic transport properties in hydrogenated graphene.
},
keywords = {Graphane, Graphene, graphone, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}

Jaques, Ygor M.; Brunetto, Gustavo; Galvao, Douglas S.
Nanodroplets Impacting on Graphene Online
2016, ((ArXiv preprint)).
Abstract | Links | BibTeX | Tags: Droplet, Graphene, Molecular Dynamics
@online{Jaques2016,
title = {Nanodroplets Impacting on Graphene},
author = {Jaques, Ygor M. and Brunetto, Gustavo and Galvao, Douglas S.},
url = {http://arxiv.org/abs/1602.02013},
year = {2016},
date = {2016-02-05},
abstract = {The unique and remarkable properties of graphene can be exploited as the basis to a wide
range of applications. However, in spite of years of investigations there are some important
graphene properties that are not still fully understood, as for example, its wettability. There are
controversial reported results whether graphene is really hydrophobic or hydrophilic. In order to
address this problem we have carried out classical molecular dynamics simulations of water
nanodroplets shot against graphene surface. Our results show that the contact angle values
between the nanodroplets and graphene surfaces depend on the initial droplet velocity value and
these angles can change from 86º (hydrophobic) to 35º (hydrophilic). Our preliminary results
indicate that the graphene wettability can be dependent on spreading liquid dynamics and which
can explain some of the apparent inconsistencies reported in the literature.},
note = {(ArXiv preprint)},
keywords = {Droplet, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}
range of applications. However, in spite of years of investigations there are some important
graphene properties that are not still fully understood, as for example, its wettability. There are
controversial reported results whether graphene is really hydrophobic or hydrophilic. In order to
address this problem we have carried out classical molecular dynamics simulations of water
nanodroplets shot against graphene surface. Our results show that the contact angle values
between the nanodroplets and graphene surfaces depend on the initial droplet velocity value and
these angles can change from 86º (hydrophobic) to 35º (hydrophilic). Our preliminary results
indicate that the graphene wettability can be dependent on spreading liquid dynamics and which
can explain some of the apparent inconsistencies reported in the literature.
Woellner, Cristiano Francisco; Autreto, Pedro Alves da Silva; Galvao, Douglas S
One Side-Graphene Hydrogenation (Graphone): Substrate Effects Online
2016, visited: 18.01.2016, ((ArXiv preprint)).
Abstract | Links | BibTeX | Tags: Graphane, Graphene, graphone, Molecular Dynamics
@online{Woellner2016,
title = {One Side-Graphene Hydrogenation (Graphone): Substrate Effects},
author = {Woellner, Cristiano Francisco and Autreto, Pedro Alves da Silva and Galvao, Douglas S},
url = {http://arxiv.org/abs/1601.04484},
year = {2016},
date = {2016-01-18},
urldate = {2016-01-18},
abstract = {Recent studies on graphene hydrogenation processes showed that hydrogenation occurs
via island growing domains, however how the substrate can affect the hydrogenation dynamics
and/or pattern formation has not been yet properly investigated. In this work we have addressed
these issues through fully atomistic reactive molecular dynamics simulations. We investigated
the structural and dynamical aspects of the hydrogenation of graphene membranes (one-side
hydrogenation, the so called graphone structure) on different substrates (graphene, few-layers
graphene, graphite and platinum). Our results also show that the observed hydrogenation rates
are very sensitive to the substrate type. For all investigated cases, the largest fraction of
hydrogenated carbon atoms was for platinum substrates. Our results also show that a significant
number of randomly distributed H clusters are formed during the early stages of the
hydrogenation process, regardless of the type of substrate and temperature. These results suggest
that, similarly to graphane formation, large perfect graphone-like domains are unlikely to be
formed. These findings are especially important since experiments have showed that cluster
formation influences the electronic transport properties in hydrogenated graphene.},
note = {(ArXiv preprint)},
keywords = {Graphane, Graphene, graphone, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}
via island growing domains, however how the substrate can affect the hydrogenation dynamics
and/or pattern formation has not been yet properly investigated. In this work we have addressed
these issues through fully atomistic reactive molecular dynamics simulations. We investigated
the structural and dynamical aspects of the hydrogenation of graphene membranes (one-side
hydrogenation, the so called graphone structure) on different substrates (graphene, few-layers
graphene, graphite and platinum). Our results also show that the observed hydrogenation rates
are very sensitive to the substrate type. For all investigated cases, the largest fraction of
hydrogenated carbon atoms was for platinum substrates. Our results also show that a significant
number of randomly distributed H clusters are formed during the early stages of the
hydrogenation process, regardless of the type of substrate and temperature. These results suggest
that, similarly to graphane formation, large perfect graphone-like domains are unlikely to be
formed. These findings are especially important since experiments have showed that cluster
formation influences the electronic transport properties in hydrogenated graphene.
2015
Andrei V Alaferdov Victor A Ermakov, Alfredo R Vaz
Burning Graphene Layer-by-Layer Journal Article
In: Nature Scientific Reports, vol. 5, pp. 11546, 2015.
Abstract | Links | BibTeX | Tags: Burning, Graphene, Molecular Dynamics, TEM
@article{Ermakov2015,
title = {Burning Graphene Layer-by-Layer},
author = {Victor A Ermakov, Andrei V Alaferdov, Alfredo R Vaz, Eric Perim, Pedro AS Autreto, Ricardo Paupitz, Douglas S Galvao, Stanislav A Moshkalev},
url = {http://www.nature.com/articles/srep11546?WT.ec_id=SREP-639-20150630},
doi = {10.1038/srep11546},
year = {2015},
date = {2015-06-23},
journal = {Nature Scientific Reports},
volume = {5},
pages = {11546},
abstract = {Graphene, in single layer or multi-layer forms, holds great promise for future electronics and high-temperature applications. Resistance to oxidation, an important property for high-temperature applications, has not yet been extensively investigated. Controlled thinning of multi-layer graphene (MLG), e.g., by plasma or laser processing is another challenge, since the existing methods produce non-uniform thinning or introduce undesirable defects in the basal plane. We report here that heating to extremely high temperatures (exceeding 2000 K) and controllable layer-by-layer burning (thinning) can be achieved by low-power laser processing of suspended high-quality MLG in air in “cold-wall” reactor configuration. In contrast, localized laser heating of supported samples results in non-uniform graphene burning at much higher rates. Fully atomistic molecular dynamics simulations were also performed to reveal details of oxidation mechanisms leading to uniform layer-by-layer graphene gasification. The extraordinary resistance of MLG to oxidation paves the way to novel high-temperature applications as continuum light source or scaffolding material.},
keywords = {Burning, Graphene, Molecular Dynamics, TEM},
pubstate = {published},
tppubtype = {article}
}
2014

Perim, E; Fonseca, AF; Pugno, NM; Galvao, DS
Violation of the universal behavior of membranes inside cylindrical tubes at nanoscale Journal Article
In: EPL (Europhysics Letters), vol. 105, no. 5, pp. 56002, 2014.
Abstract | Links | BibTeX | Tags: Graphene, Nanoscale Effects, Scrolls
@article{perim2014violation,
title = {Violation of the universal behavior of membranes inside cylindrical tubes at nanoscale},
author = {Perim, E and Fonseca, AF and Pugno, NM and Galvao, DS},
url = {http://iopscience.iop.org/0295-5075/105/5/56002},
year = {2014},
date = {2014-01-01},
journal = {EPL (Europhysics Letters)},
volume = {105},
number = {5},
pages = {56002},
publisher = {IOP Publishing},
abstract = {Recently, it was proposed based on classical elasticity theory and experiments at macroscale, that the conformations of sheets inside cylindrical tubes present a universal behavior. A natural question is whether this behavior still holds at nanoscale. Based on molecular-dynamics simulations and analytical modeling for graphene and boron nitride membranes confined inside carbon nanotubes, we show that the class of universality observed at macroscale is violated at nanoscale. The precise origin of these discrepancies is addressed and proven to be related to both surface and atomistic effects.
},
keywords = {Graphene, Nanoscale Effects, Scrolls},
pubstate = {published},
tppubtype = {article}
}
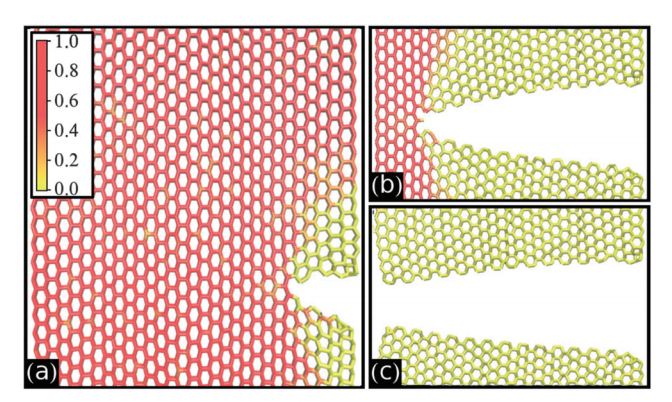
Botari, T; Perim, E; Autreto, PAS; van Duin, ACT; Paupitz, R; Galvao, DS
Mechanical properties and fracture dynamics of silicene membranes Journal Article
In: PHYSICAL CHEMISTRY CHEMICAL PHYSICS, vol. 16, no. 36, pp. 19417–19423, 2014.
Abstract | Links | BibTeX | Tags: Fracture, Germanene, Graphene, Mechanical Properties, Silicene
@article{botari2014mechanical,
title = {Mechanical properties and fracture dynamics of silicene membranes},
author = {Botari, T and Perim, E and Autreto, PAS and van Duin, ACT and Paupitz, R and Galvao, DS},
url = {http://pubs.rsc.org/en/content/articlehtml/2014/cp/c4cp02902j},
year = {2014},
date = {2014-01-01},
journal = {PHYSICAL CHEMISTRY CHEMICAL PHYSICS},
volume = {16},
number = {36},
pages = {19417--19423},
publisher = {ROYAL SOC CHEMISTRY},
abstract = {As graphene has become one of the most important materials, there is renewed interest in other similar structures. One example is silicene, the silicon analogue of graphene. It shares some of the remarkable graphene properties, such as the Dirac cone, but presents some distinct ones, such as a pronounced structural buckling. We have investigated, through density functional based tight-binding (DFTB), as well as reactive molecular dynamics (using ReaxFF), the mechanical properties of suspended single-layer silicene. We calculated the elastic constants, analyzed the fracture patterns and edge reconstructions. We also addressed the stress distributions, unbuckling mechanisms and the fracture dependence on the temperature. We analysed the differences due to distinct edge morphologies, namely zigzag and armchair.},
keywords = {Fracture, Germanene, Graphene, Mechanical Properties, Silicene},
pubstate = {published},
tppubtype = {article}
}
Autreto, PAS; de Sousa, JM; Galvao, DS
Site-dependent hydrogenation on graphdiyne Journal Article
In: Carbon, vol. 77, pp. 829–834, 2014.
Abstract | Links | BibTeX | Tags: Functionalization, Graphdyine, Graphene, Graphynes
@article{autreto2014site,
title = {Site-dependent hydrogenation on graphdiyne},
author = {Autreto, PAS and de Sousa, JM and Galvao, DS},
url = {http://www.sciencedirect.com/science/article/pii/S0008622314005429},
year = {2014},
date = {2014-01-01},
journal = {Carbon},
volume = {77},
pages = {829--834},
publisher = {Pergamon},
abstract = {Graphene is one of the most important materials in science today due to its unique and remarkable electronic, thermal and mechanical properties. However in its pristine state, graphene is a gapless semiconductor, what limits its use in transistor electronics. In part due to the revolution created by graphene in materials science, there is a renewed interest in other possible graphene-like two-dimensional structures. Examples of these structures are graphynes and graphdiynes, which are two-dimensional structures, composed of carbon atoms in sp2 and sp-hybridized states. Graphdiynes (benzenoid rings connecting two acetylenic groups) were recently synthesized and some of them are intrinsically nonzero gap systems. These systems can be easily hydrogenated and the relative level of hydrogenation can be used to tune the band gap values. We have investigated, using fully reactive molecular dynamics (ReaxFF), the structural and dynamics aspects of the hydrogenation mechanisms of graphdiyne membranes. Our results showed that the hydrogen bindings have different atom incorporation rates and that the hydrogenation patterns change in time in a very complex way. The formation of correlated domains reported to hydrogenated graphene is no longer observed in graphdiyne cases.},
keywords = {Functionalization, Graphdyine, Graphene, Graphynes},
pubstate = {published},
tppubtype = {article}
}
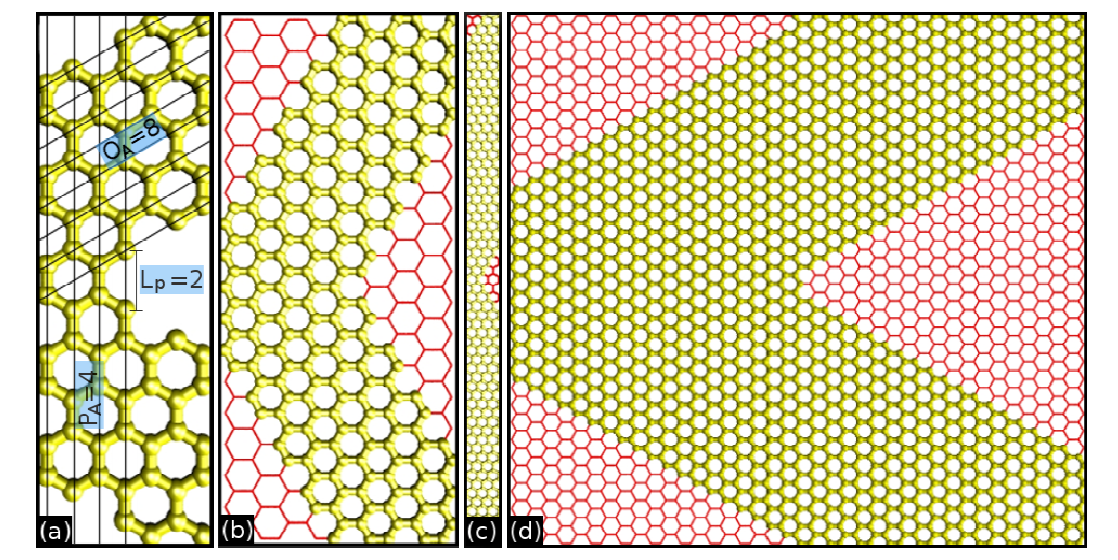
Bizao, RA; Botari, T; Galvao, DS
Mechanical Properties of Graphene Nanowiggles Proceedings
Cambridge University Press, vol. 1658, 2014.
Abstract | Links | BibTeX | Tags: Graphene, Molecular Dynamics, NanoRibbons, Nanowiggles
@proceedings{bizao2014mechanical,
title = {Mechanical Properties of Graphene Nanowiggles},
author = {Bizao, RA and Botari, T and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=9248042&fileId=S1946427414004023},
year = {2014},
date = {2014-01-01},
journal = {MRS Proceedings},
volume = {1658},
pages = {mrsf13--1658},
publisher = {Cambridge University Press},
abstract = {In this work we have investigated the mechanical properties and fracture patterns of some graphene nanowiggles (GNWs). Graphene nanoribbons are finite graphene segments with a large aspect ratio, while GNWs are nonaligned periodic repetitions of graphene nanoribbons. We have carried out fully atomistic molecular dynamics simulations using a reactive force field (ReaxFF), as implemented in the LAMPPS (Large-scale Atomic/Molecular Massively Parallel Simulator) code. Our results showed that the GNW fracture patterns are strongly dependent on the nanoribbon topology and present an interesting behavior, since some narrow sheets have larger ultimate failure strain values. This can be explained by the fact that narrow nanoribbons have more angular freedom when compared to wider ones, which can create a more efficient way to accumulate and to dissipate strain/stress. We have also observed the formation of linear atomic chains (LACs) and some structural defect reconstructions during the material rupture. The reported graphene failure patterns, where zigzag/armchair edge terminated graphene structures are fractured along armchair/zigzag lines, were not observed in the GNW analyzed cases.},
keywords = {Graphene, Molecular Dynamics, NanoRibbons, Nanowiggles},
pubstate = {published},
tppubtype = {proceedings}
}
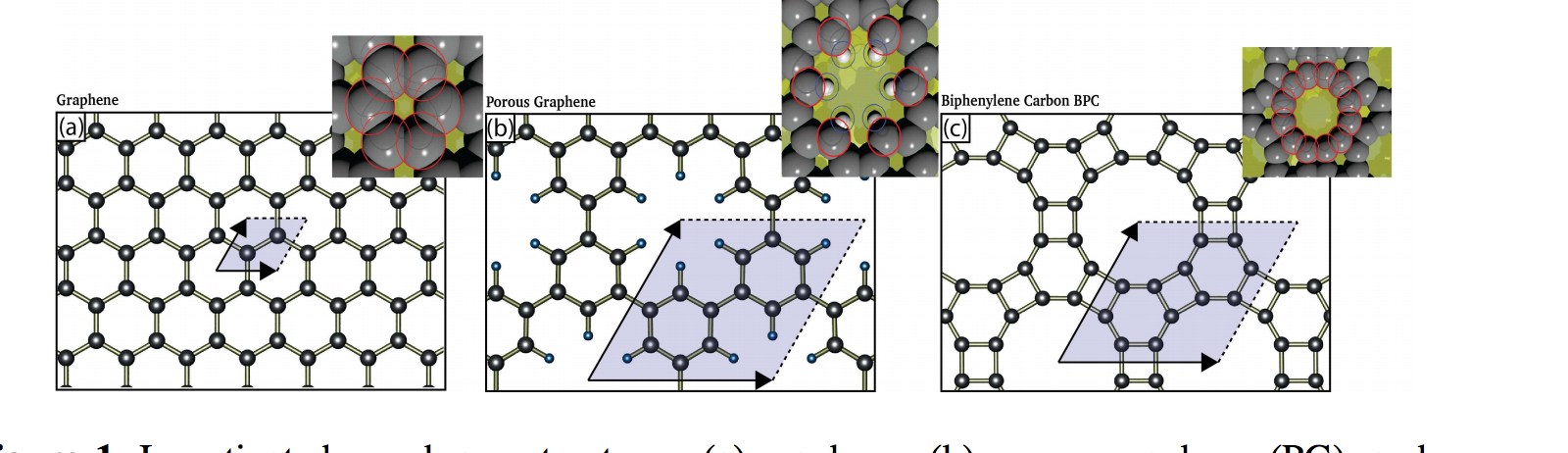
Brunetto, G; Galvao, DS
Graphene-like Membranes: From Impermeable to Selective Sieves Proceedings
Cambridge University Press, vol. 1658, 2014.
Abstract | Links | BibTeX | Tags: Graphene, Membranes, Porous Graphene, Sieves
@proceedings{brunetto2014graphene,
title = {Graphene-like Membranes: From Impermeable to Selective Sieves},
author = {Brunetto, G and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=9248039&fileId=S1946427414004011},
year = {2014},
date = {2014-01-01},
journal = {MRS Proceedings},
volume = {1658},
pages = {mrsf13--1658},
publisher = {Cambridge University Press},
abstract = {Recently, it was proposed that graphene membranes could act as impermeable atomic
structures to standard gases. For some other applications, a higher level of porosity is needed,
and the so-called Porous Graphene (PG) and Biphenylene Carbon (BPC) membranes are good
candidates to effectively work as selective sieves. In this work we have used classical molecular
dynamics simulations to study the dynamics of membrane permeation of He and Ar atoms and
possible selectivity effects. For the graphene membranes we did not observe any leakage
through the membrane and/or membrane/substrate interface until a critical pressure limit, then a
sudden membrane detachment occurs. PG and BPC membranes are not impermeable as
graphene ones, but there are significant energy barriers to diffusion depending on the atom type.
Our results show that this kind of porous membranes can be effectively used as selective sieves
for pure and mixtures of gases.},
keywords = {Graphene, Membranes, Porous Graphene, Sieves},
pubstate = {published},
tppubtype = {proceedings}
}
structures to standard gases. For some other applications, a higher level of porosity is needed,
and the so-called Porous Graphene (PG) and Biphenylene Carbon (BPC) membranes are good
candidates to effectively work as selective sieves. In this work we have used classical molecular
dynamics simulations to study the dynamics of membrane permeation of He and Ar atoms and
possible selectivity effects. For the graphene membranes we did not observe any leakage
through the membrane and/or membrane/substrate interface until a critical pressure limit, then a
sudden membrane detachment occurs. PG and BPC membranes are not impermeable as
graphene ones, but there are significant energy barriers to diffusion depending on the atom type.
Our results show that this kind of porous membranes can be effectively used as selective sieves
for pure and mixtures of gases.

Vinod, Soumya; Tiwary, Chandra Sekhar; da Silva Autreto, Pedro Alves; Taha-Tijerina, Jaime; Ozden, Sehmus; Chipara, Alin Cristian; Vajtai, Robert; Galvao, Douglas S; Narayanan, Tharangattu N; Ajayan, Pulickel M
Low-density three-dimensional foam using self-reinforced hybrid two-dimensional atomic layers Journal Article
In: Nature Communications, vol. 5, 2014.
Links | BibTeX | Tags: foams, Fracture, Graphene, Mechanical Properties, top20
@article{vinod2014low,
title = {Low-density three-dimensional foam using self-reinforced hybrid two-dimensional atomic layers},
author = {Vinod, Soumya and Tiwary, Chandra Sekhar and da Silva Autreto, Pedro Alves and Taha-Tijerina, Jaime and Ozden, Sehmus and Chipara, Alin Cristian and Vajtai, Robert and Galvao, Douglas S and Narayanan, Tharangattu N and Ajayan, Pulickel M},
url = {http://www.nature.com/ncomms/2014/140729/ncomms5541/full/ncomms5541.html},
year = {2014},
date = {2014-01-01},
journal = {Nature Communications},
volume = {5},
publisher = {Nature Publishing Group},
keywords = {foams, Fracture, Graphene, Mechanical Properties, top20},
pubstate = {published},
tppubtype = {article}
}
2013

Perim, Eric; Paupitz, Ricardo; Galvao, Douglas S
Controlled route to the fabrication of carbon and boron nitride nanoscrolls: A molecular dynamics investigation Journal Article
In: Journal of Applied Physics, vol. 113, no. 5, pp. 054306, 2013.
Abstract | Links | BibTeX | Tags: Boron Nitride, Carbon Nanotubes, Graphene, Molecular Dynamics, Scrolls
@article{perim2013controlled,
title = {Controlled route to the fabrication of carbon and boron nitride nanoscrolls: A molecular dynamics investigation},
author = {Perim, Eric and Paupitz, Ricardo and Galvao, Douglas S},
url = {http://scitation.aip.org/content/aip/journal/jap/113/5/10.1063/1.4790304},
year = {2013},
date = {2013-01-01},
journal = {Journal of Applied Physics},
volume = {113},
number = {5},
pages = {054306},
publisher = {AIP Publishing},
abstract = {Carbon nanoscrolls (graphene layers rolled up into papyrus-like tubular structures) are nanostructures with unique and interesting characteristics that could be exploited to build several new nanodevices. However, an efficient and controlled synthesis of these structures was not achieved yet, making its large scale production a challenge to materials scientists. Also, the formation process and detailed mechanisms that occur during its synthesis are not completely known. In this work, using fully atomistic molecular dynamics simulations, we discuss a possible route to nanoscrolls made from graphene layers deposited over silicon oxide substrates containing chambers/pits. The scrolling mechanism is triggered by carbon nanotubes deposited on the layers. The process is completely general and can be used to produce scrolls from other lamellar materials, like boron nitride, for instance.},
keywords = {Boron Nitride, Carbon Nanotubes, Graphene, Molecular Dynamics, Scrolls},
pubstate = {published},
tppubtype = {article}
}
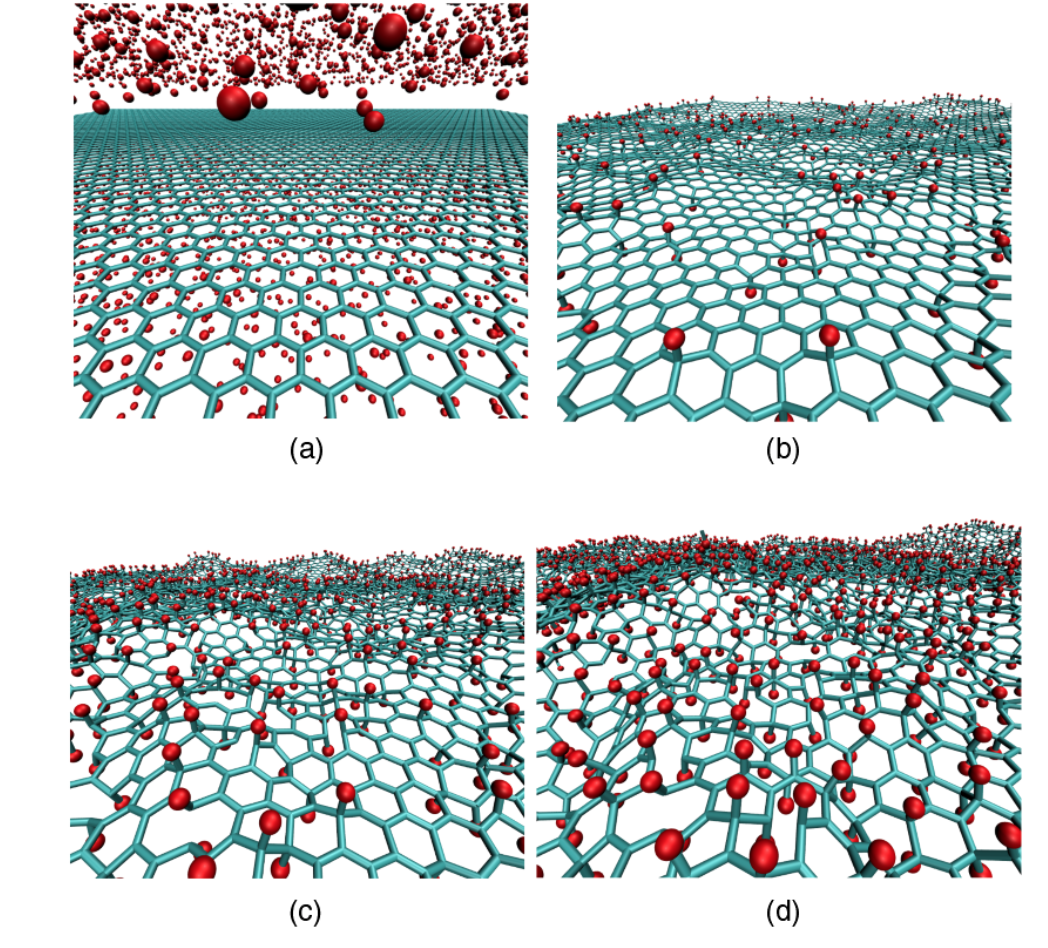
Paupitz, R; Autreto, Pedro AS; Legoas, SB; Srinivasan, S Goverapet; van Duin, Adri CT; Galvao, DS
Graphene to fluorographene and fluorographane: a theoretical study Journal Article
In: Nanotechnology, vol. 24, no. 3, pp. 035706, 2013.
Abstract | Links | BibTeX | Tags: Fluorographene, Graphane, Graphene
@article{paupitz2013graphene,
title = {Graphene to fluorographene and fluorographane: a theoretical study},
author = {Paupitz, R and Autreto, Pedro AS and Legoas, SB and Srinivasan, S Goverapet and van Duin, Adri CT and Galvao, DS},
url = {http://iopscience.iop.org/0957-4484/24/3/035706},
year = {2013},
date = {2013-01-01},
journal = {Nanotechnology},
volume = {24},
number = {3},
pages = {035706},
publisher = {IOP Publishing},
abstract = {We report here a fully reactive molecular dynamics study on the structural and dynamical aspects of the fluorination of graphene membranes (fluorographene). Our results show that fluorination tends to produce defective areas on the graphene membranes with significant distortions of carbon–carbon bonds. Depending on the amount of incorporated fluorine atoms, large membrane holes were observed due to carbon atom losses. These results may explain the broad distribution of the structural lattice parameter values experimentally observed. We have also investigated the effects of mixing hydrogen and fluorine atoms on the graphene functionalization. Our results show that, when in small amounts, the presence of hydrogen atoms produces a significant decrease in the rate of fluorine incorporation onto the membrane. On the other hand, when fluorine is the minority element, it produces a significant catalytic effect on the rate of hydrogen incorporation. We have also observed the spontaneous formation of new hybrid structures with different stable configurations (chair-like, zigzag-like and boat-like) which we named fluorographane.},
keywords = {Fluorographene, Graphane, Graphene},
pubstate = {published},
tppubtype = {article}
}
2012
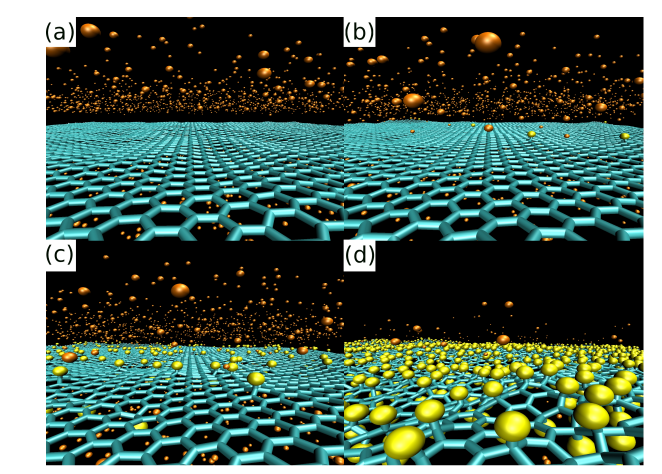
Autreto, PAS; Galvao, Douglas S; Santos, Ricardo PB; Legoas, SB
Graphene to Fluorographene: A Reactive Molecular Dynamics Study Journal Article
In: Physicæ Proceedings, vol. 1, no. 1, pp. 3, 2012.
Abstract | Links | BibTeX | Tags: Graphanes, Graphene, Molecular Dynamics
@article{autreto2012graphene,
title = {Graphene to Fluorographene: A Reactive Molecular Dynamics Study},
author = {Autreto, PAS and Galvao, Douglas S and Santos, Ricardo PB and Legoas, SB},
url = {http://physicae.ifi.unicamp.br/phyproceedings/article/view/physicae.proceedings.XIYRM.11},
year = {2012},
date = {2012-01-01},
journal = {Physicæ Proceedings},
volume = {1},
number = {1},
pages = {3},
abstract = {We have investigated, using fully reactive molecular dynamics methodology, the structural and dynamical aspects of the fluorination of graphene membranes leading to fluographene formation. The strong and fast chemical reactivity processes involving fluorine produce distinct aspects of the observed in the case of the hydrogenation of graphene (the so called graphane formation). Fluorination tends to produce significant defective areas on the graphene membrane with alteration on the typical carbon-carbon distances, sometimes with the presence of large holes due to carbon losses. This may explain the broad distribution of values of lattice parameter experimentally observed.
},
keywords = {Graphanes, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Brunetto, Gustavo; Autreto, PAS; Machado, Leonardo Dantas; Santos, BI; dos Santos, Ricardo PB; Galvao, Douglas S
Nonzero gap two-dimensional carbon allotrope from porous graphene Journal Article
In: The Journal of Physical Chemistry C, vol. 116, no. 23, pp. 12810–12813, 2012.
Abstract | Links | BibTeX | Tags: BPC, DFT, Graphene, Porous Graphene
@article{brunetto2012nonzero,
title = {Nonzero gap two-dimensional carbon allotrope from porous graphene},
author = {Brunetto, Gustavo and Autreto, PAS and Machado, Leonardo Dantas and Santos, BI and dos Santos, Ricardo PB and Galvao, Douglas S},
url = {http://pubs.acs.org/doi/abs/10.1021/jp211300n},
year = {2012},
date = {2012-01-01},
journal = {The Journal of Physical Chemistry C},
volume = {116},
number = {23},
pages = {12810--12813},
publisher = {American Chemical Society},
abstract = {Graphene is considered one of the most promising materials for future electronics. However, in its pristine form, graphene is a gapless material, which imposes limitations to its use in some electronic applications. To solve this problem, many approaches have been tried, such as physical and chemical functionalizations. These processes compromise some of the desirable graphene properties. In this work, based on ab initio quantum molecular dynamics, we showed that a two-dimensional carbon allotrope, named biphenylene carbon (BPC), can be obtained from selective dehydrogenation of porous graphene. BPC presents a nonzero bandgap and well-delocalized frontier orbitals. Synthetic routes to BPC are also addressed.},
keywords = {BPC, DFT, Graphene, Porous Graphene},
pubstate = {published},
tppubtype = {article}
}

dos Santos, Ricardo P; Machado, Leonardo D; Legoas, Sergio B; Galvao, Douglas S
Tribological Properties of Graphene and Boron-Nitride Layers: A Fully Atomistic Molecular Dynamics Study Proceedings
Cambridge University Press, vol. 1407, 2012.
Abstract | Links | BibTeX | Tags: Boron Nitride, Graphene, Molecular Dynamics, Tribology
@proceedings{dos2012tribological,
title = {Tribological Properties of Graphene and Boron-Nitride Layers: A Fully Atomistic Molecular Dynamics Study},
author = {dos Santos, Ricardo P and Machado, Leonardo D and Legoas, Sergio B and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8537106&fileId=S1946427412007063},
year = {2012},
date = {2012-01-01},
journal = {MRS Proceedings},
volume = {1407},
pages = {mrsf11--1407},
publisher = {Cambridge University Press},
abstract = {Graphene has been one of the most important subjects in materials science in the last years. Recently, the frictional characteristics of atomically thin sheets were experimentally investigated using atomic force microscopy (AFM). A new mechanism to explain the enhanced friction for these materials, based on elastic compliance has been proposed. Here, we have investigated the tribological properties of graphene and boron-nitride (single and multi-layers) membranes using fully atomistic molecular dynamics simulations. These simulations were carried out using classical force fields, as implemented in the Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) code. The used structural models contain typically hundreds of thousands of atoms. In order to mimic the experimental conditions, an artificial AFM tip was moved over the membranes and the tribological characteristics determined in terms of forces and energies. Our results are in good agreement with the available experimental data. They show that the observed enhanced tribological properties can be explained in terms of out-of-plane geometrical distortions and elastic waves propagation. They validate the general features of the model proposed by Lee et al. (Science 328, 76 (2010).},
keywords = {Boron Nitride, Graphene, Molecular Dynamics, Tribology},
pubstate = {published},
tppubtype = {proceedings}
}
2011
Autreto, Pedro AS; Flores, Marcelo Z; Legoas, Sergio B; Santos, Ricardo PB; Galvao, Douglas S
Cambridge University Press, vol. 1284, 2011.
Abstract | Links | BibTeX | Tags: Graphane, Graphene, Hydrogenation, Molecular Dynamics
@proceedings{autreto2011fully,
title = {A Fully Atomistic Reactive Molecular Dynamics Study on the Formation of Graphane from Graphene Hydrogenated Membranes.},
author = {Autreto, Pedro AS and Flores, Marcelo Z and Legoas, Sergio B and Santos, Ricardo PB and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8364784&fileId=S1946427411013583},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1284},
pages = {mrsf10--1284},
publisher = {Cambridge University Press},
abstract = {Using fully reactive molecular dynamics methodologies we investigated the structural and dynamical aspects of the fluorination mechanism leading to fluorographene formation from graphene membranes. Fluorination tends to produce significant defective areas on the membranes with variation on the typical carbon-carbon distances, sometimes with the presence of large holes due to carbon losses. The results obtained in our simulations are in good agreement with the broad distribution of values for the lattice parameter experimentally observed. We have also investigated mixed atmospheres composed by H and F atoms. When H is present in small quantities an expressive reduction on the rate of incorporation of fluorine was observed. On the other hand when fluorine atoms are present in small quantities in a hydrogen atmosphere, they induce an increasing on the hydrogen incorporation and the formation of locally self-organized structure of adsorbed H and F atoms.},
keywords = {Graphane, Graphene, Hydrogenation, Molecular Dynamics},
pubstate = {published},
tppubtype = {proceedings}
}
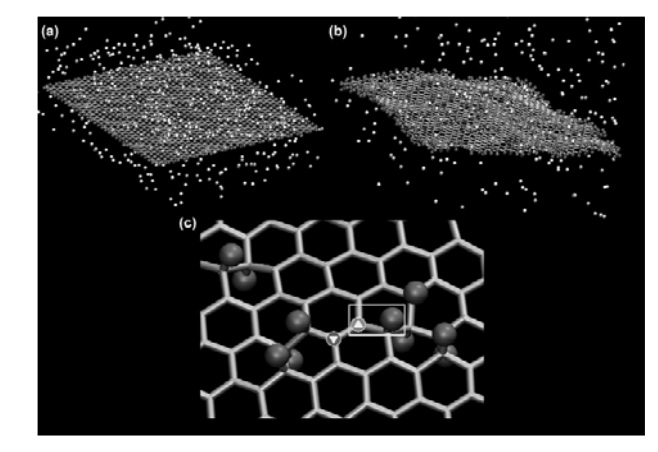
Santos, Ricardo PB; Autreto, Pedro AS; Legoas, Sergio B; Galvao, Douglas S
Cambridge University Press, vol. 1344, 2011.
Abstract | Links | BibTeX | Tags: Fluorographene, Functionalization, Graphane, Graphene
@proceedings{santos2011dynamics,
title = {The Dynamics of Formation of Graphane-like Fluorinated Graphene Membranes (Fluorographene): A Reactive Molecular Dynamics Study},
author = {Santos, Ricardo PB and Autreto, Pedro AS and Legoas, Sergio B and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayFulltext?type=1&fid=8237871&jid=OPL&volumeId=1284&issueId=-1&aid=8237869},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1344},
pages = {mrss11--1344},
publisher = {Cambridge University Press},
abstract = {Recently, Elias et al. (Science 323, 610 (2009).) reported the experimental realization of
the formation of graphane from hydrogenation of graphene membranes under cold plasma
exposure. In graphane, the carbon-carbon bonds are in sp3
configuration, as opposed to the sp2
hybridization of graphene, and the C–H bonds exhibit an alternating pattern (up and down with
relation to the plane defined by the carbon atoms). In this work we have investigated, using
reactive molecular dynamics simulations, the role of H frustration (breaking the H atoms up and
down alternating pattern) in graphane-like structures. Our results show that a significant
percentage of uncorrelated H frustrated domains are formed in the early stages of the
hydrogenation process, leading to membrane shrinkage and extensive membrane corrugations.
This might explain the significant broad distribution of values of lattice parameter
experimentally observed. For comparison purposes we have also analyzed fluorinated graphanelike
structures. Our results show that similarly to H, F atoms also create significant uncorrelated
frustrated domains on graphene membranes. },
keywords = {Fluorographene, Functionalization, Graphane, Graphene},
pubstate = {published},
tppubtype = {proceedings}
}
the formation of graphane from hydrogenation of graphene membranes under cold plasma
exposure. In graphane, the carbon-carbon bonds are in sp3
configuration, as opposed to the sp2
hybridization of graphene, and the C–H bonds exhibit an alternating pattern (up and down with
relation to the plane defined by the carbon atoms). In this work we have investigated, using
reactive molecular dynamics simulations, the role of H frustration (breaking the H atoms up and
down alternating pattern) in graphane-like structures. Our results show that a significant
percentage of uncorrelated H frustrated domains are formed in the early stages of the
hydrogenation process, leading to membrane shrinkage and extensive membrane corrugations.
This might explain the significant broad distribution of values of lattice parameter
experimentally observed. For comparison purposes we have also analyzed fluorinated graphanelike
structures. Our results show that similarly to H, F atoms also create significant uncorrelated
frustrated domains on graphene membranes.
Coutinho, Samir S; Azevedo, David L; Galvao, Douglas S
Tuning Electronic and Structural Properties of Triple Layers of Intercalated Graphene and Hexagonal Boron Nitride: An Ab-initio Study. Journal Article
In: MRS Proceedings, vol. 1307, pp. mrsf10–1307, 2011.
Abstract | Links | BibTeX | Tags: BN, DFT, Graphene, Heterostructures
@article{coutinho2011tuning,
title = {Tuning Electronic and Structural Properties of Triple Layers of Intercalated Graphene and Hexagonal Boron Nitride: An Ab-initio Study.},
author = {Coutinho, Samir S and Azevedo, David L and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8330317&fileId=S1946427411003642},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1307},
pages = {mrsf10--1307},
publisher = {Cambridge University Press},
abstract = {Recently, several experiments and theoretical studies demonstrated the possibility of tuning or modulating band gap values of nanostructures composed of bi-layer graphene, bi-layer hexagonal boron-nitride (BN) and hetero-layer combinations. These triple layers systems present several possibilities of stacking. In this work we report an ab initio (within the formalism of density functional theory (DFT)) study of structural and electronic properties of some of these stacked configurations. We observe that an applied external electric field can alter the electronic and structural properties of these systems. With the same value of the applied electric field the band gap values can be increased or decreased, depending on the layer stacking sequences. Strong geometrical deformations were observed. These results show that the application of an external electric field perpendicular to the stacked layers can effectively be used to modulate their inter-layer distances and/or their band gap values.},
keywords = {BN, DFT, Graphene, Heterostructures},
pubstate = {published},
tppubtype = {article}
}
2010
Martins, BVC; Galvao, DS
Curved graphene nanoribbons: structure and dynamics of carbon nanobelts Journal Article
In: Nanotechnology, vol. 21, no. 7, pp. 075710, 2010.
Abstract | Links | BibTeX | Tags: Folding, Graphene, Mechanical Properties, Nanobelts, NanoRibbons
@article{martins2010curved,
title = {Curved graphene nanoribbons: structure and dynamics of carbon nanobelts},
author = {Martins, BVC and Galvao, DS},
url = {http://iopscience.iop.org/0957-4484/21/7/075710},
year = {2010},
date = {2010-01-01},
journal = {Nanotechnology},
volume = {21},
number = {7},
pages = {075710},
publisher = {IOP Publishing},
abstract = {Carbon nanoribbons (CNRs) are graphene (planar) structures with a large aspect ratio. Carbon nanobelts (CNBs) are small graphene nanoribbons rolled up into spiral-like structures, i.e. carbon nanoscrolls (CNSs) with a large aspect ratio. In this work we investigated the energetics and dynamical aspects of CNBs formed from rolling up CNRs. We have carried out molecular dynamics simulations using reactive empirical bond-order potentials. Our results show that, similarly to CNSs, CNB formation is dominated by two major energy contributions, the increase in the elastic energy due to the bending of the initial planar configuration (decreasing structural stability) and the energetic gain due to van der Waals interactions of the overlapping surface of the rolled layers (increasing structural stability). Beyond a critical diameter value these scrolled structures can be even more stable (in terms of energy) than their equivalent planar configurations. In contrast to CNSs that require energy-assisted processes (sonication, chemical reactions, etc) to be formed, CNBs can be spontaneously formed from low temperature driven processes. Long CNBs (length of ~30.0 nm) tend to exhibit self-folded racket-like conformations with formation dynamics very similar to the one observed for long carbon nanotubes. Shorter CNBs will be more likely to form perfect scrolled structures. Possible synthetic routes to fabricate CNBs from graphene membranes are also addressed.
},
keywords = {Folding, Graphene, Mechanical Properties, Nanobelts, NanoRibbons},
pubstate = {published},
tppubtype = {article}
}
2009
Flores, Marcelo ZS; Autreto, Pedro AS; Legoas, Sergio B; Galvao, Douglas S
Graphene to graphane: a theoretical study Journal Article
In: Nanotechnology, vol. 20, no. 46, pp. 465704, 2009.
Abstract | Links | BibTeX | Tags: Functionalization, Graphanes, Graphene, Hydrogenation
@article{flores2009graphene,
title = {Graphene to graphane: a theoretical study},
author = {Flores, Marcelo ZS and Autreto, Pedro AS and Legoas, Sergio B and Galvao, Douglas S},
url = {http://iopscience.iop.org/0957-4484/20/46/465704},
year = {2009},
date = {2009-01-01},
journal = {Nanotechnology},
volume = {20},
number = {46},
pages = {465704},
publisher = {IOP Publishing},
abstract = {Graphane is a two-dimensional system consisting of a single layer of fully saturated (sp3 hybridization) carbon atoms. In an ideal graphane structure C–H bonds exhibit an alternating pattern (up and down with relation to the plane defined by the carbon atoms). In this work we have investigated, using ab initio and reactive molecular dynamics simulations, the role of H frustration (breaking the H atoms' up and down alternating pattern) in graphane-like structures. Our results show that a significant percentage of uncorrelated H frustrated domains are formed in the early stages of the hydrogenation process leading to membrane shrinkage and extensive membrane corrugations. These results also suggest that large domains of perfect graphane-like structures are unlikely to be formed, as H frustrated domains are always present.
},
keywords = {Functionalization, Graphanes, Graphene, Hydrogenation},
pubstate = {published},
tppubtype = {article}
}
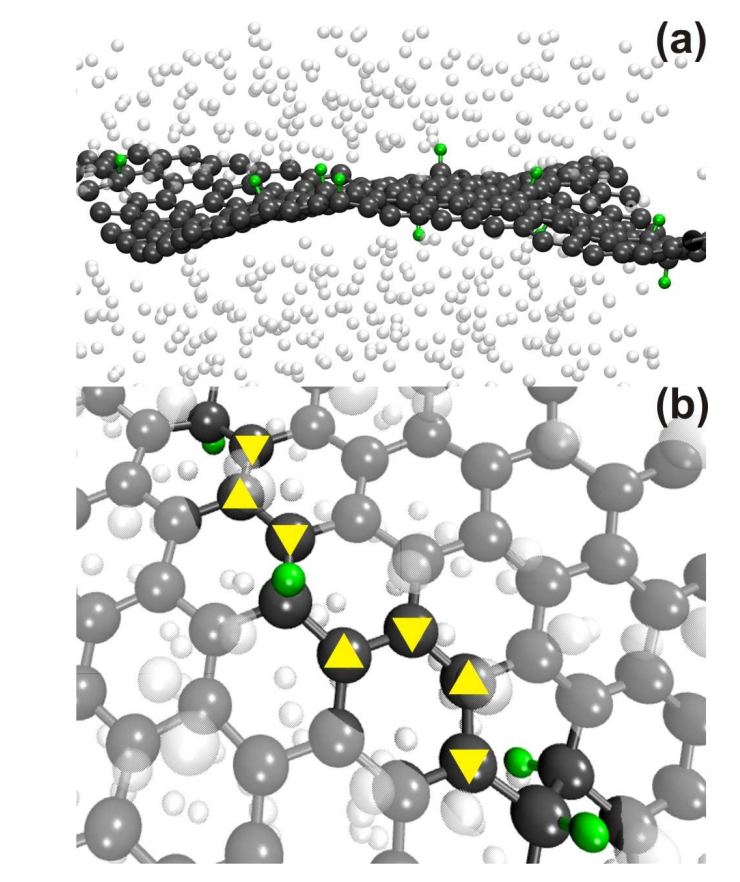
Legoas, Sergio B; Autreto, Pedro AS; Flores, Marcelo ZS; Galvao, Douglas S
Graphene to graphane: the role of H frustration in lattice contraction Journal Article
In: arXiv preprint arXiv:0903.0278, 2009.
Abstract | Links | BibTeX | Tags: Functionalization, Graphane, Graphene, Hydrogenation
@article{legoas2009graphene,
title = {Graphene to graphane: the role of H frustration in lattice contraction},
author = {Legoas, Sergio B and Autreto, Pedro AS and Flores, Marcelo ZS and Galvao, Douglas S},
url = {http://arxiv.org/abs/0903.0278},
year = {2009},
date = {2009-01-01},
journal = {arXiv preprint arXiv:0903.0278},
abstract = {Graphane is a two-dimensional system consisting of a single planar layer of fully saturated (sp3 hybridization) carbon atoms with H atoms attached to them in an alternating pattern (up and down with relation to the plane defined by the carbon atoms). Stable graphane structures were theoretically predicted to exist some years ago and just experimentally realized through hydrogenation of graphene membranes. In this work we have investigated using textit{ab initio} and reactive molecular dynamics the role of H frustration (breaking the H atoms up and down alternating pattern) in graphane-like structures. Our results show that H frustration significantly contributes to lattice contraction. The dynamical aspects of converting graphene to graphane is also addressed.},
keywords = {Functionalization, Graphane, Graphene, Hydrogenation},
pubstate = {published},
tppubtype = {article}
}
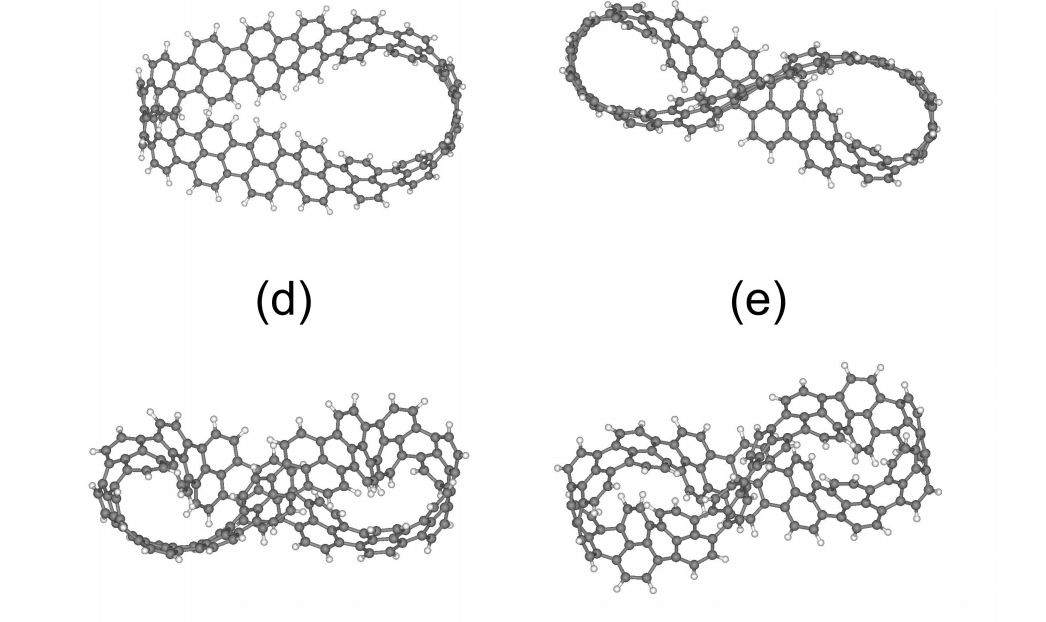
Caetano, Ewerton WS; Freire, Valder N; Santos, Sergio G dos; Galvao, Douglas S; Sato, Fernando
Mobius and twisted graphene nanoribbons: stability, geometry and electronic properties Journal Article
In: arXiv preprint arXiv:0903.2080, 2009.
Abstract | Links | BibTeX | Tags: Graphene, Mobius, NanoRibbons, Structure
@article{caetano2009m,
title = {Mobius and twisted graphene nanoribbons: stability, geometry and electronic properties},
author = {Caetano, Ewerton WS and Freire, Valder N and Santos, Sergio G dos and Galvao, Douglas S and Sato, Fernando},
url = {http://arxiv.org/abs/0903.2080},
year = {2009},
date = {2009-01-01},
journal = {arXiv preprint arXiv:0903.2080},
abstract = {Results of classical force field geometry optimizations for twisted graphene nanoribbons with a number of twists Nt varying from 0 to 7 (the case Nt=1 corresponds to a half-twist M"obius nanoribbon) are presented in this work. Their structural stability was investigated using the Brenner reactive force field. The best classical molecular geometries were used as input for semiempirical calculations, from which the electronic properties (energy levels, HOMO, LUMO orbitals) were computed for each structure. CI wavefunctions were also calculated in the complete active space framework taking into account eigenstates from HOMO-4 to LUMO+4, as well as the oscillator strengths corresponding to the first optical transitions in the UV-VIS range. The lowest energy molecules were found less symmetric than initial configurations, and the HOMO-LUMO energy gaps are larger than the value found for the nanographene used to build them due to electronic localization effects created by the twisting. A high number of twists leads to a sharp increase of the HOMO → LUMO transition energy. We suggest that some twisted nanoribbons could form crystals stabilized by dipolar interactions.},
keywords = {Graphene, Mobius, NanoRibbons, Structure},
pubstate = {published},
tppubtype = {article}
}
2008

E. W. S.; Freire Caetano, V. N. ; dos Santos
Mobius and twisted graphene nanoribbons: Stability, geometry, and electronic properties Journal Article
In: THE JOURNAL OF CHEMICAL PHYSICS, vol. 128, pp. 164719, 2008.
Abstract | Links | BibTeX | Tags: DFT, Graphene, Mobis, NanoRibbons, Structure
@article{Caetano2008,
title = {Mobius and twisted graphene nanoribbons: Stability, geometry, and electronic properties},
author = {Caetano, E. W. S.; Freire, V. N.; dos Santos, S. G.; Galvao, D. S.,and Sato, F.},
url = {http://scitation.aip.org/content/aip/journal/jcp/128/16/10.1063/1.2908739},
year = {2008},
date = {2008-04-29},
journal = {THE JOURNAL OF CHEMICAL PHYSICS},
volume = {128},
pages = {164719},
abstract = {Results of classical force field geometry optimizations for twisted graphenenanoribbons with a number of twists Nt varying from 0 to 7 (the case Nt=1 corresponds to a half-twist Möbius nanoribbon) are presented in this work. Their structural stability was investigated using the Brenner reactive force field. The best classical molecular geometries were used as input for semiempirical calculations, from which the electronic properties (energy levels, HOMO, LUMO orbitals) were computed for each structure. CI wavefunctions were also calculated in the complete active space framework taking into account eigenstates from HOMO−4 to LUMO+4, as well as the oscillator strengths corresponding to the first optical transitions in the UV-VIS range. The lowest energy molecules were found less symmetric than initial configurations, and the HOMO-LUMO energy gaps are larger than the value found for the nanographene used to build them due to electronic localization effects created by the twisting. A high number of twists leads to a sharp increase of the HOMO→LUMO transition energy. We suggest that some twisted nanoribbons could form crystals stabilized by dipolar interactions.},
keywords = {DFT, Graphene, Mobis, NanoRibbons, Structure},
pubstate = {published},
tppubtype = {article}
}
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ

