
Jaques, Ygor M; Galvao, Douglas S
Permeation of Water Nanodroplets on Carbon Nanotubes Forests Journal Article
In: MRS Advances, vol. 2017, pp. 123-128, 2017.
@article{Jaques2017b,
title = {Permeation of Water Nanodroplets on Carbon Nanotubes Forests},
author = {Jaques, Ygor M and Galvao, Douglas S},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/permeation-of-water-nanodroplets-on-carbon-nanotubes-forests/99C67F3DC0AD10DB1A4580CC8CEFDF58},
doi = {10.1557/adv.2017.129},
year = {2017},
date = {2017-01-31},
journal = {MRS Advances},
volume = {2017},
pages = {123-128},
abstract = {Fully atomistic molecular dynamics simulations were carried out to investigate how a liquid-like water droplet behaves when into contact with a nanopore formed by carbon nanotube arrays. We have considered different tube arrays, varying the spacing between them, as well as, different chemical functionalizations on the uncapped nanotubes. Our results show that simple functionalizations (for instance, hydrogen ones) allow tuning up the wetting surface properties increasing the permeation of liquid inside the nanopore. For functionalizations that increase the surface hydrophilicity, even when the pore size is significantly increased the droplet remains at the surface without tube permeation.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Jaques, Ygor M; Galvao, Douglas S
Nanodroplets Behavior on Graphdiyne Membranes Journal Article
In: MRS Advances, vol. 2017, pp. 1-6, 2017.
@article{Jaques2017,
title = {Nanodroplets Behavior on Graphdiyne Membranes},
author = {Jaques, Ygor M and Galvao, Douglas S},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/nanodroplets-behavior-on-graphdiyne-membranes/16AD56CAD07570E7F4F194A56E9680C3},
doi = {10.1557/adv.2017.128},
year = {2017},
date = {2017-01-30},
journal = {MRS Advances},
volume = {2017},
pages = {1-6},
abstract = {In this work we have investigated, by fully atomistic reactive (force field ReaxFF) molecular dynamics simulations, some aspects of impact dynamics of water nanodroplets on graphdiyne-like membranes. We simulated graphdiyne-supported membranes impacted by nanodroplets at different velocities (from 100 up to 1500 m/s). The results show that due to the graphdiyne porous and elastic structure, the droplets present an impact dynamics very complex in relation to the ones observed for graphene membranes. Under impact the droplets spread over the surface with a maximum contact radius proportional to the impact velocity. Depending on the energy impact value, a number of water molecules were able to percolate the nanopore sheets. However, even in these cases the droplet shape is preserved and the main differences between the different impact velocities cases reside on the splashing pattern at the maximum spreading.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Bizao, Rafael A; Machado, Leonardo D; de Sousa, Jose M; Pugno, Nicola M; Galvao, Douglas S
Scale Effects on the Ballistic Penetration of Graphene Sheets Online
2017, (preprint arXiv:1701.07367).
@online{Bizao2017c,
title = {Scale Effects on the Ballistic Penetration of Graphene Sheets},
author = {Bizao, Rafael A and Machado, Leonardo D and de Sousa, Jose M and Pugno, Nicola M and Galvao, Douglas S},
url = {https://arxiv.org/pdf/1701.07367.pdf},
year = {2017},
date = {2017-01-25},
abstract = {Carbon nanostructures are promising ballistic protection materials,
due to their low density and excellent mechanical properties. Recent
experimental and computational investigations on the behavior
of graphene under impact conditions revealed exceptional energy absorption
properties as well. However, the reported numerical and experimental
values differ by an order of magnitude. In this work, we
combined numerical and analytical modeling to address this issue. In
the numerical part, we employed reactive molecular dynamics to carry
out ballistic tests on single and double-layered graphene sheets. We
used velocity values within the range tested in experiments. Our numerical
and the experimental results were used to determine parameters
for a scaling law, which is in good agreement with all experimental
and simulation results. We find that the specific penetration energy
decreases as the number of layers (N) increases, from ∼ 25 MJ/kg for
N = 1 to ∼ 0.26 MJ/kg as N → ∞. These scale effects explain the
apparent discrepancy between simulations and experiments.},
note = {preprint arXiv:1701.07367},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
due to their low density and excellent mechanical properties. Recent
experimental and computational investigations on the behavior
of graphene under impact conditions revealed exceptional energy absorption
properties as well. However, the reported numerical and experimental
values differ by an order of magnitude. In this work, we
combined numerical and analytical modeling to address this issue. In
the numerical part, we employed reactive molecular dynamics to carry
out ballistic tests on single and double-layered graphene sheets. We
used velocity values within the range tested in experiments. Our numerical
and the experimental results were used to determine parameters
for a scaling law, which is in good agreement with all experimental
and simulation results. We find that the specific penetration energy
decreases as the number of layers (N) increases, from ∼ 25 MJ/kg for
N = 1 to ∼ 0.26 MJ/kg as N → ∞. These scale effects explain the
apparent discrepancy between simulations and experiments.
Peter Samora Owuor Alin Cristian Chipara, Sanjit Bhowmick
Structural Reinforcement through Liquid Encapsulation Journal Article
In: Advanced Materials Interfaces, vol. 4, pp. 1600781, 2017.
@article{Chipara2017,
title = {Structural Reinforcement through Liquid Encapsulation},
author = {Alin Cristian Chipara, Peter Samora Owuor, Sanjit Bhowmick, Gustavo Brunetto, SA Asif, Mircea Chipara, Robert Vajtai, Jun Lou, Douglas S Galvao, Chandra Sekhar Tiwary, Pulickel M Ajayan},
url = {http://onlinelibrary.wiley.com/doi/10.1002/admi.201600781/full},
doi = {10.1002/admi.201600781},
year = {2017},
date = {2017-01-23},
journal = {Advanced Materials Interfaces},
volume = {4},
pages = {1600781},
abstract = {The liquid inside a solid material is one of the most common composite materials in nature. The interface between solid–liquid plays an important role in unique deformation. Here, model systems of two polymers (polydimethylsiloxane–polyvinylidenefluoride) are used to make sphere of solid with liquid inside it.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Oliveira, Eliezer Fernando; Pedro Alves da Silva Autreto,; Galvao, Douglas Soares
Silver Hardening via Hypersonic Impacts Online
2017, (preprint arXiv:1801.04780).
@online{Oliveira2017,
title = {Silver Hardening via Hypersonic Impacts},
author = {Eliezer Fernando Oliveira and Pedro Alves da Silva Autreto, and Douglas Soares Galvao},
url = {https://arxiv.org/abs/1801.04780},
year = {2017},
date = {2017-01-15},
abstract = {The search for new ultra strong materials has been a very active research area. With relation
to metals, a successful way to improve their strength is by the creation of a gradient of
nanograins (GNG) inside the material. Recently, R. Thevamaran et al. [Science v354, 312-
316 (2016)] propose a single step method based on high velocity impact of silver nanocubes
to produce high-quality GNG. This method consists of producing high impact collisions of
silver cubes at hypersonic velocity (~400 m/s) against a rigid wall. Although they observed an
improvement in the mechanical properties of the silver after the impact, the GNG creation
and the strengthening mechanism at nanoscale remain unclear. In order to gain further
insights about these mechanisms, we carried out fully atomistic molecular dynamics
simulations (MD) to investigate the atomic conformations/rearrangements during and after
high impact collisions of silver nanocubes at ultrasonic velocity. Our results indicate the coexistence
of polycrystalline arrangements after the impact formed by core HCP domains
surrounded by FCC ones, which could also contribute to explain the structural hardening.},
note = {preprint arXiv:1801.04780},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
to metals, a successful way to improve their strength is by the creation of a gradient of
nanograins (GNG) inside the material. Recently, R. Thevamaran et al. [Science v354, 312-
316 (2016)] propose a single step method based on high velocity impact of silver nanocubes
to produce high-quality GNG. This method consists of producing high impact collisions of
silver cubes at hypersonic velocity (~400 m/s) against a rigid wall. Although they observed an
improvement in the mechanical properties of the silver after the impact, the GNG creation
and the strengthening mechanism at nanoscale remain unclear. In order to gain further
insights about these mechanisms, we carried out fully atomistic molecular dynamics
simulations (MD) to investigate the atomic conformations/rearrangements during and after
high impact collisions of silver nanocubes at ultrasonic velocity. Our results indicate the coexistence
of polycrystalline arrangements after the impact formed by core HCP domains
surrounded by FCC ones, which could also contribute to explain the structural hardening.
Alves, Ana Paula P; Koizumi, Ryota; Samanta, Atanu; Machado, Leonardo D; Singh, Abhisek K; Galvao, Douglas S; Silva, Glaura G; Tiwary, Chandra S; Ajayan, Pulickel M
One-step electrodeposited 3D-ternary composite of zirconia nanoparticles, rGO and polypyrrole with enhanced supercapacitor performance Journal Article
In: Nano Energy, vol. 31, pp. 225-232, 2017.
@article{Alves2017,
title = {One-step electrodeposited 3D-ternary composite of zirconia nanoparticles, rGO and polypyrrole with enhanced supercapacitor performance},
author = {Alves, Ana Paula P and Koizumi, Ryota and Samanta, Atanu and Machado, Leonardo D and Singh, Abhisek K and Galvao, Douglas S and Silva, Glaura G and Tiwary, Chandra S and Ajayan, Pulickel M},
url = {http://www.sciencedirect.com/science/article/pii/S221128551630502X},
doi = {10.1016/j.nanoen.2016.11.018},
year = {2017},
date = {2017-01-01},
journal = {Nano Energy},
volume = {31},
pages = {225-232},
abstract = {Supercapacitor electrodes consisting of conjugated polymers (CP), metal oxides and graphene nanosheets have been explored as a strategy to achieve high specific capacitance, power, energy density, and stability. In this work, we synthesized a 3D structure composed of zirconia oxide nanoparticles (ZrO2), reduced graphene oxide (rGO) and polypyrrole (PPy), using a simple and easily scalable one-step chronopotentiometry method. Detailed characterization revealed that the addition of rGO and ZrO2 modified the morphology of the electrode material. The capacitance of the resulting architecture improved by up to a 100%. The ternary composite featured high stability, with an increase of 5% in capacitance after a thousand cycles. DFT and MD simulations were carried out in order to provide further insight on the role of zirconia.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Chandra Sekhar Tiwary Sujin P Jose, Suppanat Kosolwattana
Enhanced supercapacitor performance of a 3D architecture tailored using atomically thin rGO–MoS 2 2D sheets Journal Article
In: RSC Advances, vol. 6, pp. 93384-93393, 2016.
@article{Jose2016,
title = {Enhanced supercapacitor performance of a 3D architecture tailored using atomically thin rGO–MoS 2 2D sheets},
author = {Sujin P Jose, Chandra Sekhar Tiwary, Suppanat Kosolwattana, Prasanth Raghavan, Leonardo D Machado, Chandkiram Gautam, T Prasankumar, Jarin Joyner, Sehmus Ozden, Douglas S Galvao, PM Ajayan},
url = {xlink.rsc.org/?DOI=c6ra20960b},
doi = {10.1039/C6RA20960B},
year = {2016},
date = {2016-09-19},
journal = {RSC Advances},
volume = {6},
pages = {93384-93393},
abstract = {A 3D architecture is fabricated using 2D nano-sheets of GO and MoS2 as the building blocks by a facile, one-pot chronoamperometry method to achieve a conductive additive free, binder free and scalable supercapacitor electrode. The superior electrochemical properties of the 3D PPy-rGO–MoS2 (PGMo) are due to its porous structure, thin wall, high surface area and high electrical conductivity that endow rapid transportation of electrolyte ions and electrons throughout the electrode matrix. The synergistic effect between the components in a proper ratio improves the supercapacitor performance and material stability of PGMo. The possible correlation of the structure and electrochemical performance of the 3D ternary composite is backed by a fully atomistic molecular dynamics (MD) simulation study. The high specific capacitance (387 F g−1) and impressive cycling stability (>1000 cycles) estimated for PGMo open up an opportunity to consider the 3D ternary nanostructures as cutting edge materials for energy storage solutions.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
P. M. Gautam, Chandkiram; Tiwary, Chandra Sekhar; Machado, Leonardo D.; Jose, Sujin; Ozden, Sehmus; Biradar, Santoshkumar; Galvao, Douglas S.; Sonker, Rakesh K.; Yadav, B. C.; Vajtai, Robert; Ajayan,
Synthesis and porous h-BN 3D architectures for effective humidity and gas sensors Authors Journal Article
In: RSC Advances, vol. 6, no. 91, pp. 87888-87896, 2016.
@article{Gautam2016,
title = {Synthesis and porous h-BN 3D architectures for effective humidity and gas sensors Authors},
author = {P. M. Gautam, Chandkiram and Tiwary, Chandra Sekhar and Machado, Leonardo D. and Jose, Sujin and Ozden, Sehmus and Biradar, Santoshkumar and Galvao, Douglas S. and Sonker, Rakesh K. and Yadav, B. C. and Vajtai, Robert and Ajayan},
url = {pubs.rsc.org/en/Content/ArticleHtml/2016/RA/c6ra18833h},
doi = {10.1039/C6RA18833H},
year = {2016},
date = {2016-09-09},
journal = {RSC Advances},
volume = {6},
number = {91},
pages = {87888-87896},
abstract = {3D (three dimensional) architectures synthesised using an easily scalable solid state method which results in an interconnected network of porous h-BN sheets with boron trioxide are reported in this study. The boron trioxide acts as a nucleating agent for the formation of laterally large nanosheets of h-BN with a low density and increases the specific surface area. The stable form shows improved mechanical properties (experimentally and using MD simulation) and serves as a suitable material for humidity and liquefied petroleum gas (LPG) sensor applications. The sensor shows stability for up to several months without losing its sensitivity.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Leonardo D Machado Sehmus Ozden, ChandraSekhar Tiwary
Ballistic Fracturing of Carbon Nanotubes Journal Article
In: ACS Applied Materials & Interfaces, vol. 8, no. 37, pp. 24819-24825, 2016.
@article{Ozden2016b,
title = {Ballistic Fracturing of Carbon Nanotubes},
author = {Sehmus Ozden, Leonardo D Machado, ChandraSekhar Tiwary, Pedro AS Autreto, Robert Vajtai, Enrique V Barrera, Douglas S Galvao, Pulickel M Ajayan},
url = {pubs.acs.org/doi/abs/10.1021/acsami.6b07547},
doi = {10.1021/acsami.6b07547},
year = {2016},
date = {2016-09-08},
journal = {ACS Applied Materials & Interfaces},
volume = {8},
number = {37},
pages = {24819-24825},
abstract = {Advanced materials with multifunctional capabilities and high resistance to hypervelocity impact are of great interest to the designers of aerospace structures. Carbon nanotubes (CNTs) with their lightweight and high strength properties are alternative to metals and/or metallic alloys conventionally used in aerospace applications. Here we report a detailed study on the ballistic fracturing of CNTs for different velocity ranges. Our results show that the highly energetic impacts cause bond breakage and carbon atom rehybridizations, and sometimes extensive structural reconstructions were also observed. Experimental observations show the formation of nanoribbons, nanodiamonds, and covalently interconnected nanostructures, depending on impact conditions. Fully atomistic reactive molecular dynamics simulations were also carried out in order to gain further insights into the mechanism behind the transformation of CNTs. The simulations show that the velocity and relative orientation of the multiple colliding nanotubes are critical to determine the impact outcome.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Chandra Sekhar Tiwary Mohamad A Kabbani, Anirban Som
A generic approach for mechano-chemical reactions between carbon nanotubes of different functionalities Journal Article
In: Carbon, vol. 104, pp. 196-202, 2016.
@article{Kabbani2016,
title = {A generic approach for mechano-chemical reactions between carbon nanotubes of different functionalities},
author = {Mohamad A Kabbani, Chandra Sekhar Tiwary, Anirban Som, KR Krishnadas, Pedro AS Autreto, Sehmus Ozden, Kunttal Keyshar, Ken Hackenberg, Alin Christian Chipara, Douglas S Galvao, Robert Vajtai, Ahmad T Kabbani, Thalappil Pradeep, Pulickel M Ajayan},
url = {www.sciencedirect.com/science/article/pii/S000862231630183X},
doi = {10.1016/j.carbon.2016.02.094},
year = {2016},
date = {2016-08-31},
journal = {Carbon},
volume = {104},
pages = {196-202},
abstract = {Abstract Here, we report similar reactions between nanotubes carrying functionalities,
namely carbon nanotubes (CNTs) with the acyl chloride/hydroxyl and amine/carboxylic
functionalities directly attached to their surfaces, resulting in the formation ofchemically
modified graphene products. The reaction is spontaneous and is facilitated by simple
grinding of the reactants. The new solid-state reactions have been confirmed using different
spectroscopic and electron microscopy techniques.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
namely carbon nanotubes (CNTs) with the acyl chloride/hydroxyl and amine/carboxylic
functionalities directly attached to their surfaces, resulting in the formation ofchemically
modified graphene products. The reaction is spontaneous and is facilitated by simple
grinding of the reactants. The new solid-state reactions have been confirmed using different
spectroscopic and electron microscopy techniques.
Chandra Sekhar Tiwary Dibyendu Chakravarty, Cristano F Woellner
3D Porous Graphene by Low-Temperature Plasma Welding for Bone Implants Journal Article
In: Advanced Materials, vol. 28, no. 40, pp. 8959-8967, 2016.
@article{chakravarty20163d,
title = {3D Porous Graphene by Low-Temperature Plasma Welding for Bone Implants},
author = {Dibyendu Chakravarty, Chandra Sekhar Tiwary, Cristano F Woellner, Sruthi Radhakrishnan, Soumya Vinod, Sehmus Ozden, Pedro Alves da Silva Autreto, Sanjit Bhowmick, Syed Asif, Sendurai A Mani, Douglas S Galvao, Pulickel M},
url = {onlinelibrary.wiley.com/doi/10.1002/adma.201603146/abstract },
doi = {10.1002/adma.201603146},
year = {2016},
date = {2016-08-26},
journal = {Advanced Materials},
volume = {28},
number = {40},
pages = {8959-8967},
abstract = {3D scaffolds of graphene, possessing ultra-low density, macroporous microstructure, and high yield strength and stiffness can be developed by a novel plasma welding process. The bonding between adjacent graphene sheets is investigated by molecular dynamics simulations. The high degree of biocompatibility along with high porosity and good mechanical properties makes graphene an ideal material for use as body implants.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Yongji Gong Bo Li, Zhili Hu
Solid–Vapor Reaction Growth of Transition‐Metal Dichalcogenide Monolayers Journal Article
In: Angewandte Chemie, vol. 128, no. 36, pp. 10814-10819, 2016.
@article{Li2016,
title = {Solid–Vapor Reaction Growth of Transition‐Metal Dichalcogenide Monolayers},
author = {Bo Li, Yongji Gong, Zhili Hu, Gustavo Brunetto, Yingchao Yang, Gonglan Ye, Zhuhua Zhang, Sidong Lei, Zehua Jin, Elisabeth Bianco, Xiang Zhang, Weipeng Wang, Jun Lou, Douglas S Galvão, Ming Tang, Boris I Yakobson, Robert Vajtai, Pulickel M Ajayan},
url = {onlinelibrary.wiley.com/doi/10.1002/anie.201604445/abstract},
doi = {10.1002/ange.201604445},
year = {2016},
date = {2016-08-26},
journal = {Angewandte Chemie},
volume = {128},
number = {36},
pages = {10814-10819},
abstract = {Two-dimensional (2D) layered semiconducting transition-metal dichalcogenides (TMDCs) are promising candidates for next-generation ultrathin, flexible, and transparent electronics. Chemical vapor deposition (CVD) is a promising method for their controllable, scalable synthesis but the growth mechanism is poorly understood. Herein, we present systematic studies to understand the CVD growth mechanism of monolayer MoSe2, showing reaction pathways for growth from solid and vapor precursors. Examination of metastable nanoparticles deposited on the substrate during growth shows intermediate growth stages and conversion of non-stoichiometric nanoparticles into stoichiometric 2D MoSe2 monolayers. The growth steps involve the evaporation and reduction of MoO3 solid precursors to sub-oxides and stepwise reactions with Se vapor to finally form MoSe2. The experimental results and proposed model were corroborated by ab initio Car–Parrinello molecular dynamics studies.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Amelia HC Hart Ryota Koizumi, Gustavo Brunetto
Mechano-chemical stabilization of three-dimensional carbon nanotube aggregates Journal Article
In: Carbon, vol. 110, pp. 27-33, 2016.
@article{koizumi2016mechano,
title = {Mechano-chemical stabilization of three-dimensional carbon nanotube aggregates},
author = {Ryota Koizumi, Amelia HC Hart, Gustavo Brunetto, Sanjit Bhowmick, Peter S Owuor, John T Hamel, Anieph X Gentles, Sehmus Ozden, Jun Lou, Robert Vajtai, SA Syed Asif, Douglas S Galvão, CS Tiwary, PM Ajayan},
url = {www.sciencedirect.com/science/article/pii/S0008622316307400},
doi = {10.1016/j.carbon.2016.08.085},
year = {2016},
date = {2016-08-21},
journal = {Carbon},
volume = {110},
pages = {27-33},
abstract = {Here we report a combined study of experiments and simulations to understand how chemical functional groups can mechanically stabilize aggregates of carbon nanotubes (CNTs). Ultralow density aggregates of chemically functionalized CNTs, in the form of macro-scale spheres made by freeze-drying method, show mechanical stabilization and near complete elastic recovery during deformation. Simulations of interacting functionalized carbon nanotube aggregates show better structural retention compared to non-functionalized CNTs under compression, suggesting that the atomic-level interactions between functional groups on adjoining CNTs help maintain structural rigidity and elastic response during loading. Aggregates of non-functionalized CNTs collapses under similar loading conditions. The dynamic mechanical responses of CNT macrostructures and mechano-chemical stabilization are directly observed using in-situ deformation inside a scanning electron microscope.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Chandra Sekhar Tiwary Soumya Vinod, Leonardo D Machado
Synthesis of ultralow density 3D graphene–CNT foams using a two-step method Journal Article
In: Nanoscale, vol. 8, no. 35, pp. 15857-15863, 2016.
@article{Vinod2016b,
title = {Synthesis of ultralow density 3D graphene–CNT foams using a two-step method},
author = {Soumya Vinod, Chandra Sekhar Tiwary, Leonardo D Machado, Sehmus Ozden, Robert Vajtai, Douglas S Galvao, Pulickel M Ajayan},
url = {xlink.rsc.org/?DOI=c6nr04252j},
doi = {10.1039/C6NR04252J},
year = {2016},
date = {2016-08-09},
journal = {Nanoscale},
volume = {8},
number = {35},
pages = {15857-15863},
abstract = {Here, we report a highly scalable two-step method to produce graphene foams with ordered carbon nanotube reinforcements. In our approach, we first used solution assembly methods to obtain graphene oxide foam. Next, we employed chemical vapor deposition to simultaneously grow carbon nanotubes and thermally reduce the 3D graphene oxide scaffold. The resulting structure presented increased stiffness, good mechanical stability and oil absorption properties. Molecular dynamics simulations were carried out to further elucidate failure mechanisms and to understand the enhancement of the mechanical properties. The simulations showed that mechanical failure is directly associated with bending of vertical reinforcements, and that, for similar length and contact area, much more stress is required to bend the corresponding reinforcements of carbon nanotubes, thus explaining the experimentally observed enhanced mechanical properties.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
T Botari JM de Sousa, E Perim
Mechanical and structural properties of graphene-like carbon nitride sheets Journal Article
In: RSC Advances, vol. 6, no. 80, pp. 76915-76921, 2016.
@article{deSousa2016b,
title = {Mechanical and structural properties of graphene-like carbon nitride sheets},
author = {JM de Sousa, T Botari, E Perim, RA Bizao, Douglas S Galvao},
url = {pubs.rsc.org/en/content/articlehtml/2016/ra/c6ra14273g},
doi = {10.1039/C6RA14273G},
year = {2016},
date = {2016-08-08},
journal = {RSC Advances},
volume = {6},
number = {80},
pages = {76915-76921},
abstract = {Carbon nitride-based nanostructures have attracted special attention (from theory and experiments) due to their remarkable electromechanical properties. In this work we have investigated the mechanical properties of some graphene-like carbon nitride membranes through fully atomistic reactive molecular dynamics simulations. We have analyzed three different structures of these CN families, the so-called graphene-based g-CN, triazine-based g-C3N4 and heptazine-based g-C3N4. The stretching dynamics of these membranes was studied for deformations along their two main axes and at three different temperatures: 10 K, 300 K and 600 K. We show that g-CN membranes have the lowest ultimate fracture strain value, followed by heptazine-based and triazine-based ones, respectively. This behavior can be explained in terms of their differences in density values, topologies and types of chemical bonds. The dependency of the fracture patterns on the stretching directions is also discussed.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Shaoli Fang Jiangtao Di, Francisco A Moura
Strong, Twist‐Stable Carbon Nanotube Yarns and Muscles by Tension Annealing at Extreme Temperatures Journal Article
In: Advanced Materials, vol. 28, no. 31, pp. 6598-6605, 2016.
@article{Di2016,
title = {Strong, Twist‐Stable Carbon Nanotube Yarns and Muscles by Tension Annealing at Extreme Temperatures},
author = {Jiangtao Di, Shaoli Fang, Francisco A Moura, Douglas S Galvão, Julia Bykova, Ali Aliev, Mônica Jung de Andrade, Xavier Lepró, Na Li, Carter Haines, Raquel Ovalle‐Robles, Dong Qian, Ray H Baughman},
url = {onlinelibrary.wiley.com/doi/10.1002/adma.201600628/full},
doi = {10.1002/adma.201600628},
year = {2016},
date = {2016-08-01},
journal = {Advanced Materials},
volume = {28},
number = {31},
pages = {6598-6605},
abstract = {A high-speed incandescent tension annealing process (ITAP) is used to increase the modulus and strength of twist-spun carbon nanotube yarns by up to 12-fold and 2.6-fold, respectively, provide remarkable resistance to oxidation and powerful protonating acids, and freeze yarn untwist. This twist stability enables torsional artificial-muscle motors having improved performance and minimizes problematic untwist during weaving nanotube yarns.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Rodrigo Prioli Clara M Almeida, Benjamin Fragneaud
Giant and Tunable Anisotropy of Nanoscale Friction in Graphene Journal Article
In: Nature Scientific Reports, vol. 6, pp. 31569, 2016.
@article{Almeida2016,
title = {Giant and Tunable Anisotropy of Nanoscale Friction in Graphene},
author = {Clara M Almeida, Rodrigo Prioli, Benjamin Fragneaud, Luiz Gustavo Cançado, Ricardo Paupitz, Douglas S Galvão, Marcelo De Cicco, Marcos G Menezes, Carlos A Achete, Rodrigo B Capaz},
url = {http://www-nature-com.ez88.periodicos.capes.gov.br/articles/srep31569},
doi = {10.1038/srep31569},
year = {2016},
date = {2016-07-18},
journal = {Nature Scientific Reports},
volume = {6},
pages = {31569},
abstract = {The nanoscale friction between an atomic force microscopy tip and graphene is investigated using friction force microscopy (FFM). During the tip movement, friction forces are observed to increase and then saturate in a highly anisotropic manner. As a result, the friction forces in graphene are highly dependent on the scanning direction: under some conditions, the energy dissipated along the armchair direction can be 80% higher than along the zigzag direction. In comparison, for highly-oriented pyrolitic graphite (HOPG), the friction anisotropy between armchair and zigzag directions is only 15%. This giant friction anisotropy in graphene results from anisotropies in the amplitudes of flexural deformations of the graphene sheet driven by the tip movement, not present in HOPG. The effect can be seen as a novel manifestation of the classical phenomenon of Euler buckling at the nanoscale, which provides the non-linear ingredients that amplify friction anisotropy. Simulations based on a novel version of the 2D Tomlinson model (modified to include the effects of flexural deformations), as well as fully atomistic molecular dynamics simulations and first-principles density-functional theory (DFT) calculations, are able to reproduce and explain the experimental observations.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Pedro Alves da Silva Autreto Cristiano Francisco Woellner, Douglas S Galvao
Graphone (one-side hydrogenated graphene) formation on different substrates Online
2016.
@online{Woellner2016b,
title = {Graphone (one-side hydrogenated graphene) formation on different substrates},
author = {Cristiano Francisco Woellner, Pedro Alves da Silva Autreto, Douglas S Galvao},
url = {arXiv preprint arXiv:1606.09235},
year = {2016},
date = {2016-06-29},
abstract = {In this work we present a fully atomistic reactive (ReaxFF force field) molecular dynamics study of the structural and dynamical aspects of the one-side hydrogenation of graphene membranes, leading to the formation of the so-called graphone structure. We have considered different substrates: graphene, few-layers graphene, graphite and platinum at different temperatures. Our results showed that the hydrogenation rates are very dependent on the substrate and thermal effects. Our results also showed that, similarly to graphane, large hydrogenated domains are unlikely to be formed. These hydrogenation processes occur through the formation of uncorrelated cluster domains.},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Sehmus Ozden Leonardo D Machado, ChandraSekhar Tiwary
The structural and dynamical aspects of boron nitride nanotubes under high velocity impacts Journal Article
In: Physical Chemistry Chemical Physics, vol. 18, pp. 14776-14781, 2016.
@article{Machado2016,
title = {The structural and dynamical aspects of boron nitride nanotubes under high velocity impacts},
author = {Leonardo D Machado, Sehmus Ozden, ChandraSekhar Tiwary, Pedro AS Autreto, Robert Vajtai, Enrique V Barrera, Douglas S Galvao, Pulickel M Ajayan},
url = {xlink.rsc.org/?DOI=c6cp01949h},
doi = {10.1039/C6CP01949H},
year = {2016},
date = {2016-05-01},
journal = {Physical Chemistry Chemical Physics},
volume = {18},
pages = {14776-14781},
abstract = {This communication report is a study on the structural and dynamical aspects of boron nitride nanotubes (BNNTs) shot at high velocities (∼5 km s−1) against solid targets. The experimental results show unzipping of BNNTs and the formation of hBN nanoribbons. Fully atomistic reactive molecular dynamics simulations were also carried out to gain insights into the BNNT fracture patterns and deformation mechanisms. Our results show that longitudinal and axial tube fractures occur, but the formation of BN nanoribbons from fractured tubes was only observed for some impact angles. Although some structural and dynamical features of the impacts are similar to the ones reported for CNTs, because BNNTs are more brittle than CNTs this results in a larger number of fractured tubes but with fewer formed nanoribbons.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Botari, Tiago; Paupitz, Ricardo; da Silva Autreto, Pedro Alves; Galvao, Douglas S
Graphene healing mechanisms: A theoretical investigation Journal Article
In: Carbon, vol. 99, pp. 302-309, 2016.
@article{2016Healing,
title = {Graphene healing mechanisms: A theoretical investigation},
author = {Botari, Tiago and Paupitz, Ricardo and da Silva Autreto, Pedro Alves and Galvao, Douglas S},
url = {http://www.sciencedirect.com/science/article/pii/S0008622315304784},
doi = {10.1016/j.carbon.2015.11.070},
year = {2016},
date = {2016-04-30},
journal = {Carbon},
volume = {99},
pages = {302-309},
abstract = {Large holes in graphene membranes were recently shown to heal, either at room temperature during a low energy STEM experiment, or by annealing at high temperatures. However, the details of the healing mechanism remain unclear. We carried out fully atomistic reactive molecular dynamics simulations in order to address these mechanisms under different experimental conditions. Our results show that, if a carbon atom source is present, high temperatures can provide enough energy for the carbon atoms to overcome the potential energy barrier and to produce perfect reconstruction of the graphene hexagonal structure. At room temperature, this perfect healing is only possible if the heat effects of the electron beam from STEM experiment are explicitly taken into account. The reconstruction process of a perfect or near perfect graphene structure involves the formation of linear carbon chains, as well as rings containing 5, 6, 7 and 8 atoms with planar (Stone-Wales like) and non-planar (lump like) structures. These results shed light on the healing mechanism of graphene when subjected to different experimental conditions. Additionally, the methodology presented here can be useful for investigating the tailoring and manipulations of other nano-structures.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2009
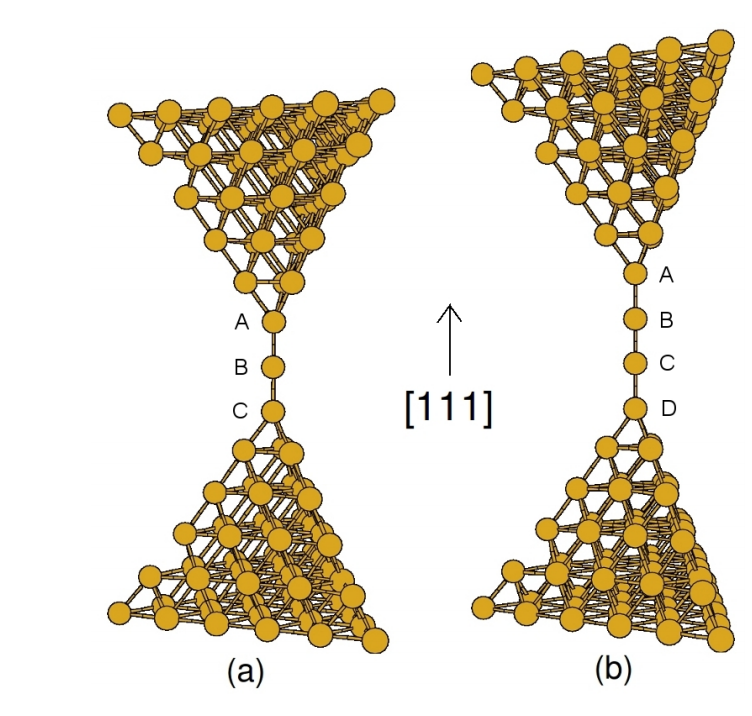
Sato, Fernando; Lagos, Maureen; Autreto, Pedro; Ugarte, Daniel; Galvao, Douglas
On the Lifetime of Suspended Atomic Chains Formed from Stretched Metallic Gold Nanowires Journal Article
In: Physicae, vol. 8, no. 8, 2009, (Invited Paper).
Abstract | Links | BibTeX | Tags: Atomic Chains, Gold, Metallic Nanowires, TEM
@article{sato2009lifetime,
title = {On the Lifetime of Suspended Atomic Chains Formed from Stretched Metallic Gold Nanowires},
author = {Sato, Fernando and Lagos, Maureen and Autreto, Pedro and Ugarte, Daniel and Galvao, Douglas},
url = {http://physicae.ifi.unicamp.br/physicae/article/viewArticle/109},
year = {2009},
date = {2009-01-01},
journal = {Physicae},
volume = {8},
number = {8},
abstract = {Metallic nanowires have been object of intense theoretical and experimental works in the lastyears. In spite of the large number of studies for such systems some fundamental aspects remain open and polemical questions. In this work we report preliminary results for the study of the final steps of Au suspended atomic chains (LACs) with different number of atoms as a function of tem-perature. We have carried out classical molecular dynamics simulations using tight-binding models with a second moment approximations. Our results suggest a more complex phenomenon than previously anticipated. The dynamics of chain rupture seems to be determined beyond thermodynamics contributions and the bond breaking patterns were observed to be chain-length dependent.
},
note = {Invited Paper},
keywords = {Atomic Chains, Gold, Metallic Nanowires, TEM},
pubstate = {published},
tppubtype = {article}
}
Torriani, IL; Silva, JC; Autreto, PAS; Galvao, DS; Caldas, MJ; Graeff, CFO
Low resolution structure of synthetic melanin aggregates in aqueous solutions and organic solvents Journal Article
In: Acta Crystalographica A, vol. 64, pp. C552, 2009.
Abstract | Links | BibTeX | Tags: Melanin, Structure
@article{torriani2009low,
title = {Low resolution structure of synthetic melanin aggregates in aqueous solutions and organic solvents},
author = {Torriani, IL and Silva, JC and Autreto, PAS and Galvao, DS and Caldas, MJ and Graeff, CFO},
url = {http://journals.iucr.org/a/issues/2008/a1/00/a39972/a39972.pdf},
year = {2009},
date = {2009-01-01},
journal = {Acta Crystalographica A},
volume = {64},
pages = {C552},
abstract = {In an effort to find out details of the melanin fundamental structural
unit, a great amount of information has been gathered using
several techniques. The local short range order of the melanin
molecular clusters has been described as consisting of five to seven
5,6-indolequinone units, arranged in planes which are pi-stacked
with a spacing of 0.34 nm. Typical cluster size is 1.5-2.0 nm in
lateral dimensions and 1.0 nm; in height. Nonetheless, structural
details and dimensions of the aggregates are still not clearly defined
and experiments did not answer the key question concerning the
identification of the fundamental melanin protomolecule. More
recently, small angle scattering of X-rays (SAXS) and neutrons
(SANS) were performed. Several authors used these techniques,
which are well designed to study macromolecules in solution to
find details of melanin-copper ions interaction as well as chemical
bleaching effects. A diversity of aggregated structures were proposed
for these nanoscaled particles based on size and apparent shape.
In this presentation we report the results of SAXS experiments
performed with melanin synthetized from L-dopa and L-tyrosine in
organic solvents, which were reported to be very effective for thin
film formation. Water-based synthetic melanin was also studied
for comparison purposes, since molecular aggregation behavior is
known to vary with the route used for the synthesis. Reliable data
was obtained for the water-based and DMSO dispersions. Data
analysis was performed by conventional IFT methods and the overall
shape and dimensional parameters of the melanin particles were
obtained. Using ab-initio calculations, a low resolution 3D model is
proposed for the basic melanin particle in aqueous media and DMSO.},
keywords = {Melanin, Structure},
pubstate = {published},
tppubtype = {article}
}
unit, a great amount of information has been gathered using
several techniques. The local short range order of the melanin
molecular clusters has been described as consisting of five to seven
5,6-indolequinone units, arranged in planes which are pi-stacked
with a spacing of 0.34 nm. Typical cluster size is 1.5-2.0 nm in
lateral dimensions and 1.0 nm; in height. Nonetheless, structural
details and dimensions of the aggregates are still not clearly defined
and experiments did not answer the key question concerning the
identification of the fundamental melanin protomolecule. More
recently, small angle scattering of X-rays (SAXS) and neutrons
(SANS) were performed. Several authors used these techniques,
which are well designed to study macromolecules in solution to
find details of melanin-copper ions interaction as well as chemical
bleaching effects. A diversity of aggregated structures were proposed
for these nanoscaled particles based on size and apparent shape.
In this presentation we report the results of SAXS experiments
performed with melanin synthetized from L-dopa and L-tyrosine in
organic solvents, which were reported to be very effective for thin
film formation. Water-based synthetic melanin was also studied
for comparison purposes, since molecular aggregation behavior is
known to vary with the route used for the synthesis. Reliable data
was obtained for the water-based and DMSO dispersions. Data
analysis was performed by conventional IFT methods and the overall
shape and dimensional parameters of the melanin particles were
obtained. Using ab-initio calculations, a low resolution 3D model is
proposed for the basic melanin particle in aqueous media and DMSO.
2008

E. W. S.; Freire Caetano, V. N. ; dos Santos
Mobius and twisted graphene nanoribbons: Stability, geometry, and electronic properties Journal Article
In: THE JOURNAL OF CHEMICAL PHYSICS, vol. 128, pp. 164719, 2008.
Abstract | Links | BibTeX | Tags: DFT, Graphene, Mobis, NanoRibbons, Structure
@article{Caetano2008,
title = {Mobius and twisted graphene nanoribbons: Stability, geometry, and electronic properties},
author = {Caetano, E. W. S.; Freire, V. N.; dos Santos, S. G.; Galvao, D. S.,and Sato, F.},
url = {http://scitation.aip.org/content/aip/journal/jcp/128/16/10.1063/1.2908739},
year = {2008},
date = {2008-04-29},
journal = {THE JOURNAL OF CHEMICAL PHYSICS},
volume = {128},
pages = {164719},
abstract = {Results of classical force field geometry optimizations for twisted graphenenanoribbons with a number of twists Nt varying from 0 to 7 (the case Nt=1 corresponds to a half-twist Möbius nanoribbon) are presented in this work. Their structural stability was investigated using the Brenner reactive force field. The best classical molecular geometries were used as input for semiempirical calculations, from which the electronic properties (energy levels, HOMO, LUMO orbitals) were computed for each structure. CI wavefunctions were also calculated in the complete active space framework taking into account eigenstates from HOMO−4 to LUMO+4, as well as the oscillator strengths corresponding to the first optical transitions in the UV-VIS range. The lowest energy molecules were found less symmetric than initial configurations, and the HOMO-LUMO energy gaps are larger than the value found for the nanographene used to build them due to electronic localization effects created by the twisting. A high number of twists leads to a sharp increase of the HOMO→LUMO transition energy. We suggest that some twisted nanoribbons could form crystals stabilized by dipolar interactions.},
keywords = {DFT, Graphene, Mobis, NanoRibbons, Structure},
pubstate = {published},
tppubtype = {article}
}
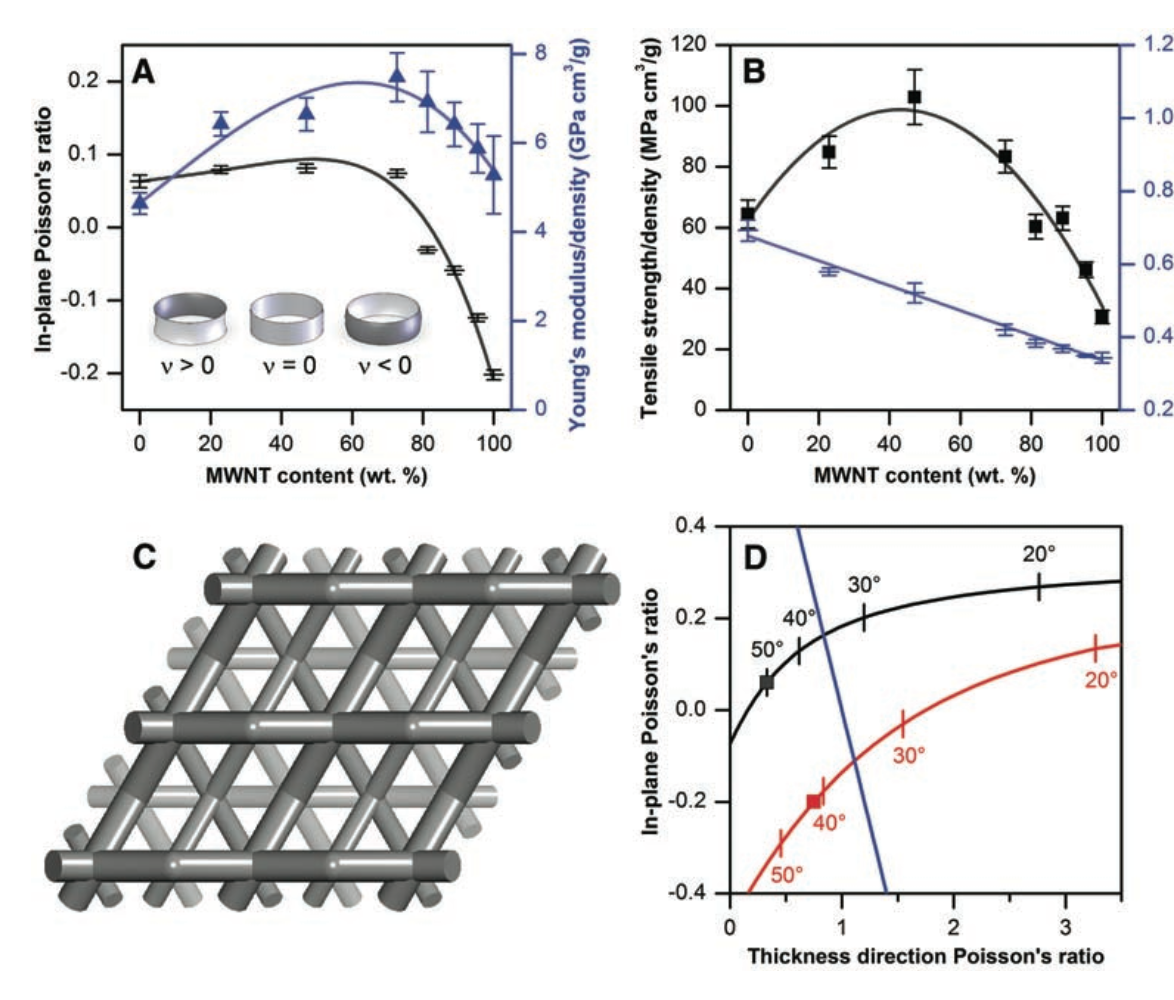
Hall, Lee J; Coluci, Vitor R; Galvao, Douglas S; Kozlov, Mikhail E; Zhang, Mei; Dantas, Socrates O; Baughman, Ray H
Sign change of Poisson's ratio for carbon nanotube sheets Journal Article
In: Science, vol. 320, no. 5875, pp. 504–507, 2008.
Abstract | Links | BibTeX | Tags: Artificial Muscles, Auxetics, Carbon Nanotube Forests, sheets, top20
@article{hall2008sign,
title = {Sign change of Poisson's ratio for carbon nanotube sheets},
author = {Hall, Lee J and Coluci, Vitor R and Galvao, Douglas S and Kozlov, Mikhail E and Zhang, Mei and Dantas, Socrates O and Baughman, Ray H},
url = {http://www.sciencemag.org/content/320/5875/504.short},
year = {2008},
date = {2008-01-01},
journal = {Science},
volume = {320},
number = {5875},
pages = {504--507},
publisher = {American Association for the Advancement of Science},
abstract = {Most materials shrink laterally like a rubber band when stretched, so their Poisson's ratios are positive. Likewise, most materials contract in all directions when hydrostatically compressed and decrease density when stretched, so they have positive linear compressibilities. We found that the in-plane Poisson's ratio of carbon nanotube sheets (buckypaper) can be tuned from positive to negative by mixing single-walled and multiwalled nanotubes. Density-normalized sheet toughness, strength, and modulus were substantially increased by this mixing. A simple model predicts the sign and magnitude of Poisson's ratio for buckypaper from the relative ease of nanofiber bending and stretch, and explains why the Poisson's ratios of ordinary writing paper are positive and much larger. Theory also explains why the negative in-plane Poisson's ratio is associated with a large positive Poisson's ratio for the sheet thickness, and predicts that hydrostatic compression can produce biaxial sheet expansion. This tunability of Poisson's ratio can be exploited in the design of sheet-derived composites, artificial muscles, gaskets, and chemical and mechanical sensors.},
keywords = {Artificial Muscles, Auxetics, Carbon Nanotube Forests, sheets, top20},
pubstate = {published},
tppubtype = {article}
}

Martins, Bruno VC; Brunetto, Gustavo; Sato, Fernando; Coluci, Vitor R; Galvao, Douglas S
Designing conducting polymers using bioinspired ant algorithms Journal Article
In: Chemical Physics Letters, vol. 453, no. 4, pp. 290–295, 2008.
Abstract | Links | BibTeX | Tags: ANTS algorithms, Artificial Intelligence, Conducting Polymer, Design of Materials
@article{martins2008designing,
title = {Designing conducting polymers using bioinspired ant algorithms},
author = {Martins, Bruno VC and Brunetto, Gustavo and Sato, Fernando and Coluci, Vitor R and Galvao, Douglas S},
url = {http://www.sciencedirect.com/science/article/pii/S0009261408000845},
year = {2008},
date = {2008-01-01},
journal = {Chemical Physics Letters},
volume = {453},
number = {4},
pages = {290--295},
publisher = {Elsevier},
abstract = {Ant algorithms are inspired in real ants and the main idea is to create virtual ants that travel into the space of possible solutions depositing virtual pheromone proportional to how good a specific solution is. This creates an autocatalytic (positive feedback) process that can be used to generate automatic solutions to very difficult problems. In the present work we show that these algorithms can be used coupled to tight-binding Hamiltonians to design conducting polymers with pre-specified properties. The methodology is completely general and can be used for a large number of optimizations problems in materials science.},
keywords = {ANTS algorithms, Artificial Intelligence, Conducting Polymer, Design of Materials},
pubstate = {published},
tppubtype = {article}
}
Coluci, Vitor R; Hall, Lee J; Kozlov, Mikhail E; Zhang, Mei; Dantas, Socrates O; Galvao, Douglas S; Baughman, Ray H
Modeling the auxetic transition for carbon nanotube sheets Journal Article
In: Physical Review B, vol. 78, no. 11, pp. 115408, 2008.
Abstract | Links | BibTeX | Tags: Auxetics, Carbon Nanotube Forests, Carbon Nanotubes, CNT sheets
@article{coluci2008modeling,
title = {Modeling the auxetic transition for carbon nanotube sheets},
author = {Coluci, Vitor R and Hall, Lee J and Kozlov, Mikhail E and Zhang, Mei and Dantas, Socrates O and Galvao, Douglas S and Baughman, Ray H},
url = {http://journals.aps.org/prb/abstract/10.1103/PhysRevB.78.115408},
year = {2008},
date = {2008-01-01},
journal = {Physical Review B},
volume = {78},
number = {11},
pages = {115408},
publisher = {APS},
abstract = {A simple model is developed to predict the complex mechanical properties of carbon nanotube sheets (buckypaper) [L. J. Hall et al., Science 320, 504 (2008)]. Fabricated using a similar method to that deployed for making writing paper, these buckypapers can have in-plane Poisson’s ratios changed from positive to negative, becoming auxetic, as multiwalled carbon nanotubes are increasingly mixed with single-walled carbon nanotubes. Essential structural features of the buckypapers are incorporated into the model: isotropic in-plane mechanical properties, nanotubes preferentially oriented in the sheet plane, and freedom to undergo stress-induced elongation by both angle and length changes. The expressions derived for the Poisson’s ratios enabled quantitative prediction of both observed properties and remarkable new properties obtainable by structural modification.},
keywords = {Auxetics, Carbon Nanotube Forests, Carbon Nanotubes, CNT sheets},
pubstate = {published},
tppubtype = {article}
}
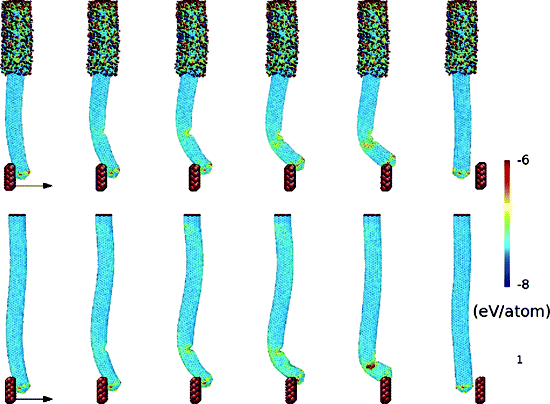
Nakabayashi, D; Moreau, ALD; Coluci, VR; Galvao, DS; Cotta, MA; Ugarte, D
Carbon nanotubes as reinforcement elements of composite nanotools Journal Article
In: Nano letters, vol. 8, no. 3, pp. 842–847, 2008.
Abstract | Links | BibTeX | Tags: AFM tips, Carbon Nanotubes, Molecular Dynamics, Nanocomposites, Tribology
@article{nakabayashi2008carbon,
title = {Carbon nanotubes as reinforcement elements of composite nanotools},
author = {Nakabayashi, D and Moreau, ALD and Coluci, VR and Galvao, DS and Cotta, MA and Ugarte, D},
url = {http://pubs.acs.org/doi/abs/10.1021/nl0729633},
year = {2008},
date = {2008-01-01},
journal = {Nano letters},
volume = {8},
number = {3},
pages = {842--847},
publisher = {American Chemical Society},
abstract = {Nanotechnology is stimulating the development of nanomanipulators, including tips to interact with individual nanosystems. Fabricating nanotips fulfilling the requirements of shape (size, aspect ratio), mechanical, magnetic, and electrical properties is a material science challenge. Here, we report the generation of reinforced carbon−carbon composite nanotools using a nanotube (CNTs) covered by an amorphous carbon matrix (shell); the CNT tip protruded and remained uncoated to preserve apex size. Unsuitable properties such as flexibility and vibration could be controlled without deteriorating the CNT size, strength, and resilience. Nanomanipulation experiments and molecular dynamics simulations have been used to study the mechanical response of these composite beams under bending efforts. AFM probes based on these C−C composite high aspect ratio tips generated excellent image resolution and showed no degradation after acquiring several hundred (400) images.},
keywords = {AFM tips, Carbon Nanotubes, Molecular Dynamics, Nanocomposites, Tribology},
pubstate = {published},
tppubtype = {article}
}
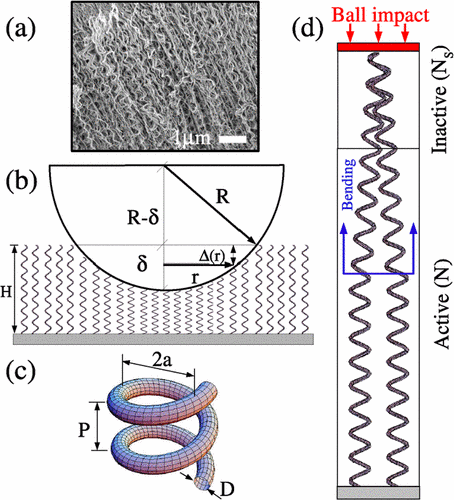
Coluci, Vitor R; Fonseca, Alexandre F; Galvao, Douglas S; Daraio, Chiara
Entanglement and the nonlinear elastic behavior of forests of coiled carbon nanotubes Journal Article
In: Physical Review Letters, vol. 100, no. 8, pp. 086807, 2008.
Abstract | Links | BibTeX | Tags: Carbon Nanotube Forests, Entanglement, Mechanical Properties, top20
@article{coluci2008entanglement,
title = {Entanglement and the nonlinear elastic behavior of forests of coiled carbon nanotubes},
author = {Coluci, Vitor R and Fonseca, Alexandre F and Galvao, Douglas S and Daraio, Chiara},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.100.086807},
year = {2008},
date = {2008-01-01},
journal = {Physical Review Letters},
volume = {100},
number = {8},
pages = {086807},
publisher = {American Physical Society},
abstract = {Helical or coiled nanostructures have been objects of intense experimental and theoretical studies due to their special electronic and mechanical properties. Recently, it was experimentally reported that the dynamical response of a foamlike forest of coiled carbon nanotubes under mechanical impact exhibits a nonlinear, non-Hertzian behavior, with no trace of plastic deformation. The physical origin of this unusual behavior is not yet fully understood. In this Letter, based on analytical models, we show that the entanglement among neighboring coils in the superior part of the forest surface must be taken into account for a full description of the strongly nonlinear behavior of the impact response of a drop ball onto a forest of coiled carbon nanotubes.},
keywords = {Carbon Nanotube Forests, Entanglement, Mechanical Properties, top20},
pubstate = {published},
tppubtype = {article}
}
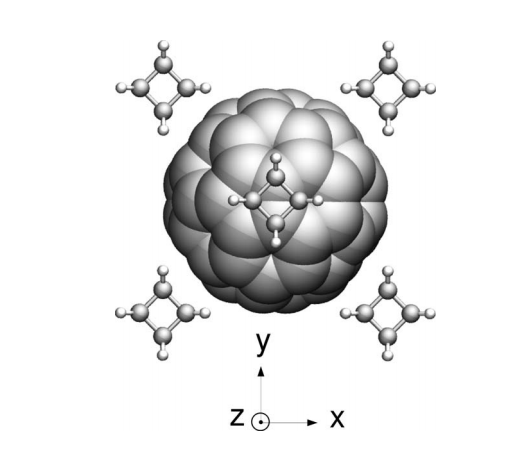
Coluci, Vitor R; Sato, Fernando; Braga, Scheila F; Skaf, Munir S; Galvao, Douglas S
Rotational dynamics and polymerization of C60 in C60-cubane crystals: A molecular dynamics study Journal Article
In: The Journal of Chemical Physics, vol. 129, no. 6, pp. 064506, 2008.
Abstract | Links | BibTeX | Tags: C60, C70, Cubanes, Fullerenes, Molecular Dynamics, Rotor-Stator
@article{coluci2008rotational,
title = {Rotational dynamics and polymerization of C60 in C60-cubane crystals: A molecular dynamics study},
author = {Coluci, Vitor R and Sato, Fernando and Braga, Scheila F and Skaf, Munir S and Galvao, Douglas S},
url = {http://scitation.aip.org/content/aip/journal/jcp/129/6/10.1063/1.2965885},
year = {2008},
date = {2008-01-01},
journal = {The Journal of Chemical Physics},
volume = {129},
number = {6},
pages = {064506},
publisher = {AIP Publishing},
abstract = {We report classical and tight-binding molecular dynamics simulations of the C60fullerene and cubane molecular crystal in order to investigate the intermolecular dynamics and polymerization processes. Our results show that, for 200 and 400 K, cubane molecules remain basically fixed, presenting only thermal vibrations, while C60fullerenes show rotational motions. Fullerenes perform “free” rotational motions at short times (≲1 ps), small amplitude hindered rotational motions (librations) at intermediate times, and rotational diffusive dynamics at long times (≳10 ps). The mechanisms underlying these dynamics are presented. Random copolymerizations among cubanes and fullerenes were observed when temperature is increased, leading to the formation of a disordered structure. Changes in the radial distribution function and electronic density of states indicate the coexistence of amorphous and crystalline phases. The different conformational phases that cubanes and fullerenes undergo during the copolymerization process are discussed.},
keywords = {C60, C70, Cubanes, Fullerenes, Molecular Dynamics, Rotor-Stator},
pubstate = {published},
tppubtype = {article}
}
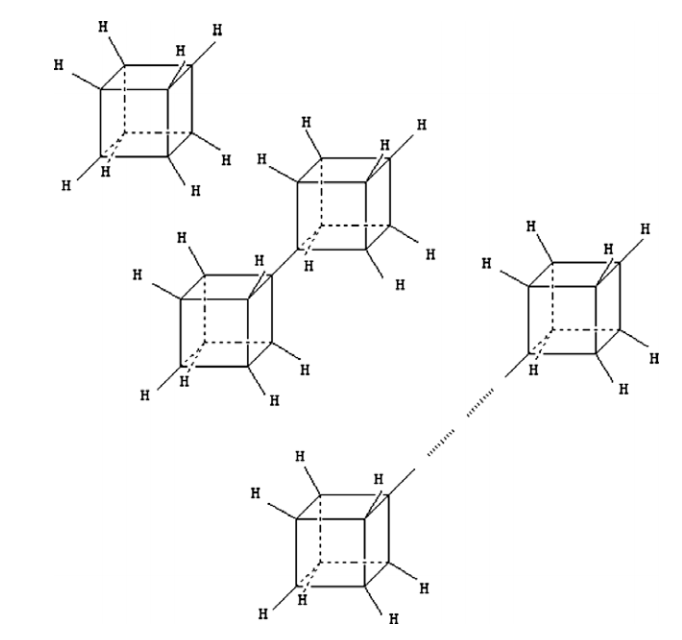
Konstantinova, Elena; Camilo Jr, Alexandre; Barone, Paulo MVB; Dantas, Socrates O; Galvao, Douglas S
Some electronic properties of saturated and unsaturated cubane oligomers using DFT-based calculations Journal Article
In: Journal of Molecular Structure: THEOCHEM, vol. 868, no. 1, pp. 37–41, 2008.
Abstract | Links | BibTeX | Tags: Cubanes, DFT, Polymer
@article{konstantinova2008some,
title = {Some electronic properties of saturated and unsaturated cubane oligomers using DFT-based calculations},
author = {Konstantinova, Elena and Camilo Jr, Alexandre and Barone, Paulo MVB and Dantas, Socrates O and Galvao, Douglas S},
url = {http://www.sciencedirect.com/science/article/pii/S016612800800448X},
year = {2008},
date = {2008-01-01},
journal = {Journal of Molecular Structure: THEOCHEM},
volume = {868},
number = {1},
pages = {37--41},
publisher = {Elsevier},
abstract = {Cubanes and cubane-based molecular structures attract considerable interest as structural units which represent a new class of materials with remarkable properties. These structures are potentially useful for a variety of industrial applications and, for this reason, deserve detailed study. One of the options is to use cubane-based structures to synthesize a new class of conducting polymers with small energy band gap. In the present work we use the DFT-based methods to perform geometrical optimization and obtain some electronic properties for cubane, cubatriene, saturated and unsaturated oligomers containing different number of cubane and cubatriene building units. Our results indicate that the energy difference between the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) manifests a small decrease with the growing units number for saturated or unsaturated oligomers. This energy difference is strongly dependent on the presence of hydrogen atoms and is greater for unsaturated structures.},
keywords = {Cubanes, DFT, Polymer},
pubstate = {published},
tppubtype = {article}
}
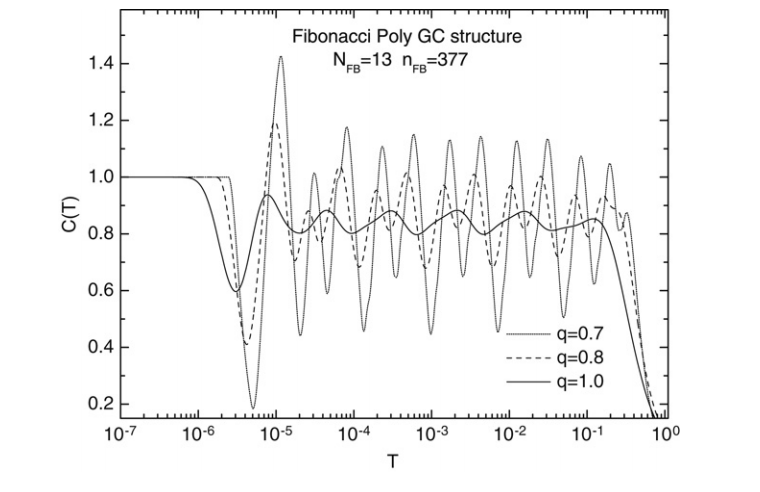
Moreira, DA; Albuquerque, EL; da Silva, LR; Galvao, DS
Low-temperature specific heat spectra considering nonextensive long-range correlated quasiperiodic DNA molecules Journal Article
In: Physica A: Statistical Mechanics and its Applications, vol. 387, no. 22, pp. 5477–5482, 2008.
Abstract | Links | BibTeX | Tags: DNA sequences, Fibonacci, nonextensive
@article{moreira2008low,
title = {Low-temperature specific heat spectra considering nonextensive long-range correlated quasiperiodic DNA molecules},
author = {Moreira, DA and Albuquerque, EL and da Silva, LR and Galvao, DS},
url = {http://www.sciencedirect.com/science/article/pii/S0378437108005347},
year = {2008},
date = {2008-01-01},
journal = {Physica A: Statistical Mechanics and its Applications},
volume = {387},
number = {22},
pages = {5477--5482},
publisher = {North-Holland},
abstract = {We consider the low-temperature specific heat spectra of long-range correlated quasiperiodic DNA molecules using a q-gaussian distribution, and compare them with those considering the Boltzmann-Gibbs distribution. The energy spectra are calculated using the one-dimensional Schrödinger equation in a tight-binding approximation with the on-site energy exhibiting long-range disorder and non-random hopping amplitudes. We focus our attention at the low temperature region, where the specific heat spectra presents a logarithmic-periodic oscillations as a function of the temperature T around a mean value given by a characteristic dimension of the energy spectrum.},
keywords = {DNA sequences, Fibonacci, nonextensive},
pubstate = {published},
tppubtype = {article}
}
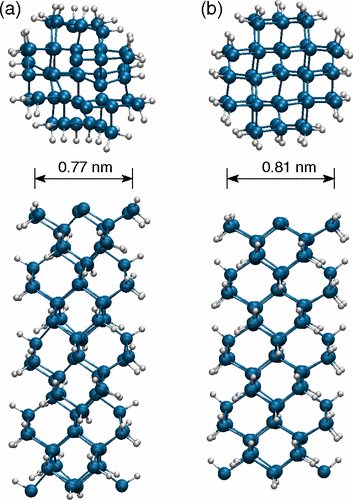
Rurali, R; Cartoixa, X; Galvao, DS
Large electromechanical response in silicon nanowires predicted from first-principles electronic structure calculations Journal Article
In: Physical Review B, vol. 77, no. 7, pp. 073403, 2008.
Abstract | Links | BibTeX | Tags: DFT, Eletroactuation, Nanowires, Silicon
@article{rurali2008large,
title = {Large electromechanical response in silicon nanowires predicted from first-principles electronic structure calculations},
author = {Rurali, R and Cartoixa, X and Galvao, DS},
url = {http://journals.aps.org/prb/abstract/10.1103/PhysRevB.77.073403},
year = {2008},
date = {2008-01-01},
journal = {Physical Review B},
volume = {77},
number = {7},
pages = {073403},
publisher = {American Physical Society},
abstract = {We study by means of first-principles electronic structure calculations the electromechanical response, i.e., the structural modifications upon charge injection, of ⟨100⟩ silicon nanowires. We show that, at variance with sp2 carbon nanostructures, the response is remarkably linear, discriminates between injected charge of different signs, and is up to one order of magnitude larger than in carbon nanotubes.
},
keywords = {DFT, Eletroactuation, Nanowires, Silicon},
pubstate = {published},
tppubtype = {article}
}
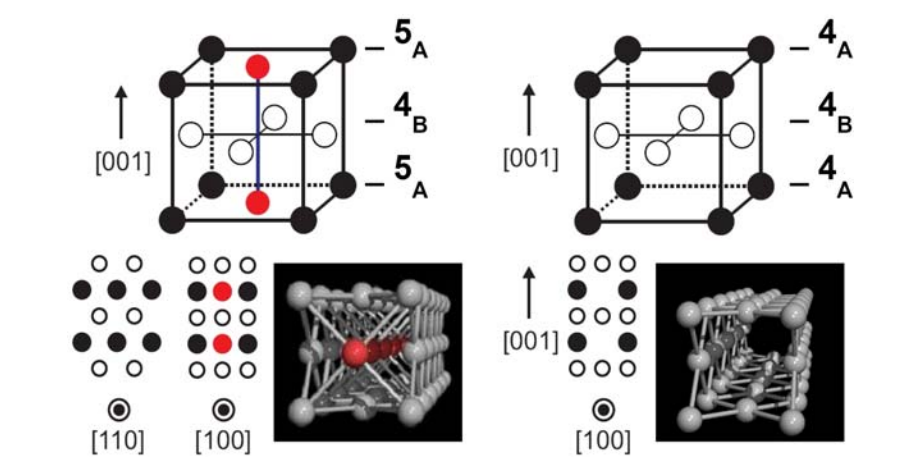
Lagos, M; Sato, F; Bettini, J; Rdrigues, V; Galvao, D; Ugarte, D
Atomic-size Silver Nanotube Book Section
In: EMC 2008 14th European Microscopy Congress 1--5 September 2008, Aachen, Germany, pp. 493–494, Springer Berlin Heidelberg, 2008, (Book Chapter).
Abstract | Links | BibTeX | Tags: Metallic Nanowires, New Structures, Silver Nanotubes, TEM
@incollection{lagos2008atomic,
title = {Atomic-size Silver Nanotube},
author = {Lagos, M and Sato, F and Bettini, J and Rdrigues, V and Galvao, D and Ugarte, D},
url = {http://link.springer.com/chapter/10.1007%2F978-3-540-85156-1_247},
year = {2008},
date = {2008-01-01},
booktitle = {EMC 2008 14th European Microscopy Congress 1--5 September 2008, Aachen, Germany},
pages = {493--494},
publisher = {Springer Berlin Heidelberg},
abstract = {The atomic arrangement of nanosystems may be quite different from the traditional materials; surface energy minimization plays a dominant role in this size range, and accounts for many of these new structures. Graphitic nanotubes [1] represent the best example, being fromed by a rolled the graphitic layer, which is tradionally flat. Subsequently the rolling of the compact (111) atomic planes was reported for gold nanowires (NW) generated by mechanical stretching [2]. But, we may expect many more surprises from the interplay between atomic and electronic structure.},
note = {Book Chapter},
keywords = {Metallic Nanowires, New Structures, Silver Nanotubes, TEM},
pubstate = {published},
tppubtype = {incollection}
}
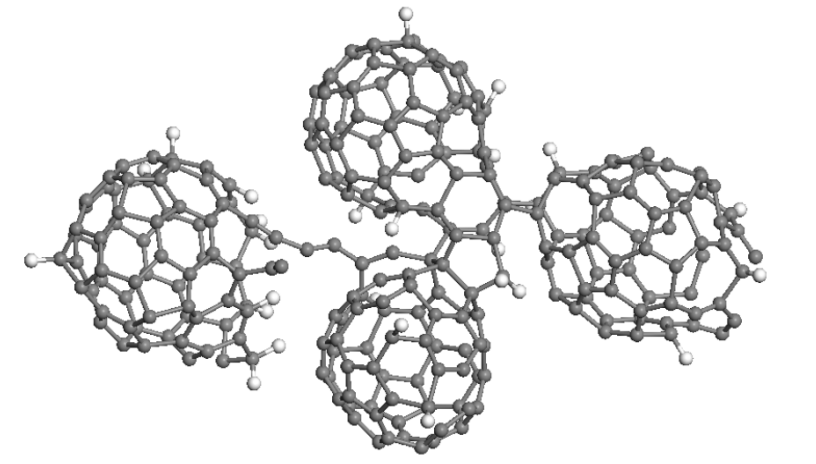
Coluci, Vitor; Sato, Fernando; Braga, Scheila F; Skaf, Munir S; Galvao, Douglas S
A molecular dynamics study of the rotational dynamics and polymerization of C60 in C60-cubane crystals Journal Article
In: MRS Proceedings, vol. 1130, pp. 1130–W06, 2008.
Abstract | Links | BibTeX | Tags: Cubanes, Molecular Dynamics, Molecular Machines, Rotor-Stator
@article{coluci2008molecular,
title = {A molecular dynamics study of the rotational dynamics and polymerization of C60 in C60-cubane crystals},
author = {Coluci, Vitor and Sato, Fernando and Braga, Scheila F and Skaf, Munir S and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=7973166&fileId=S1946427400024088},
year = {2008},
date = {2008-01-01},
journal = {MRS Proceedings},
volume = {1130},
pages = {1130--W06},
publisher = {Cambridge University Press},
abstract = {Recently, heteromolecular crystals of fullerene C60 and cubane (C8H8) have been synthesized. For some temperatures the C60 molecules are free to rotate whereas cubanes behave like a static bearing in a so-called rotor-stator phases. In this work we report classical and tight-binding molecular dynamics simulations in order to investigate the rotor-stator dynamics and polymerization processes. Our results show that, for 200 K and 400 K, cubane molecules remain basically fixed, presenting only thermal vibrations within the timescale of our simulations, while C60 fullerenes show rotational motions. Fullerenes perform “free” rotational motions at short times (< 1 ps), small amplitude hindered rotational motions (librations) at intermediate times, and rotational diffusive dynamics at long times (> 10 ps). Random copolymerization among cubanes and fullerenes were observed when temperature is increased, leading to the formation of a disordered structure.},
keywords = {Cubanes, Molecular Dynamics, Molecular Machines, Rotor-Stator},
pubstate = {published},
tppubtype = {article}
}
Nakabayashi, D; Ugarte, D; Moreau, ALD; Coluci, VR; Galvao, DS; Cotta, MA
Carbon nanotubes as R-bars of high aspect ratio composite nanotools Technical Report
2008.
Abstract | Links | BibTeX | Tags: AFM tips, Carbon Nanotubes, Nanocomposites, Tribology
@techreport{nakabayashi2008carbonb,
title = {Carbon nanotubes as R-bars of high aspect ratio composite nanotools},
author = {Nakabayashi, D and Ugarte, D and Moreau, ALD and Coluci, VR and Galvao, DS and Cotta, MA},
url = {http://lnls.cnpem.br/ar2008/},
year = {2008},
date = {2008-01-01},
abstract = {Nano technology requires the development of nano scale tools to manipulate nano systems. From the point of view of materials science, this represents a serious challenge, because nano tools must meet a series of stringent requirements of shape (size, aspect ratio), mechanical, magnetic and electrical properties. We have developed long and narrow carbon-carbon composite nano tips using carbon nanotubes covered by an amorphous carbon shell; the very small nano tube tip remained uncoated to preserve apex size. This configuration renders the system stiffer and allows for the control of flexibility and vibrations. In addition, we have maintained the important nano tube properties of size, strength and resilience. Nano manipulation experiments in situ in a high resolution scanning electron microscope were used to optimize the tips behavior and molecular dynamics simulations were used to study the mechanical response. Finally, we performed a practical application in atomic force microscopy. Composite tips yielded excellent image resolution and showed remarkable wear resistance (no degradation of image quality after acquiring several hundred images). },
keywords = {AFM tips, Carbon Nanotubes, Nanocomposites, Tribology},
pubstate = {published},
tppubtype = {techreport}
}
2007
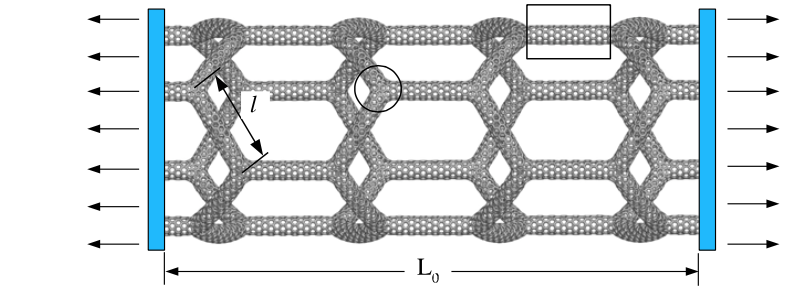
Coluci, Vitor R; Pugno, Nicola M; Dantas, Socrates O; Galvao, Douglas S; Jorio, Ado
Atomistic simulations of the mechanical properties of'super'carbon nanotubes Journal Article
In: Nanotechnology, vol. 18, no. 33, pp. 335702, 2007.
Abstract | Links | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, Super Carbons
@article{coluci2007atomistic,
title = {Atomistic simulations of the mechanical properties of'super'carbon nanotubes},
author = {Coluci, Vitor R and Pugno, Nicola M and Dantas, Socrates O and Galvao, Douglas S and Jorio, Ado},
url = {http://iopscience.iop.org/0957-4484/18/33/335702
},
year = {2007},
date = {2007-01-01},
journal = {Nanotechnology},
volume = {18},
number = {33},
pages = {335702},
publisher = {IOP Publishing},
abstract = {The mechanical properties of the so-called 'super' carbon nanotubes (STs) are investigated using classical molecular dynamics simulations. The STs are built from single-walled carbon nanotubes (SWCNTs) connected by Y-like junctions forming an ordered carbon nanotube network that is then rolled into a seamless cylinder. We observed that the ST behaviour under tensile tests is similar to the one presented by fishing nets. This interesting behaviour provides a way to vary the accessible channels to the inner parts of STs by applying an external mechanical load. The Young's modulus is dependent on the ST chirality and it inversely varies with the ST radius. Smaller reduction of breaking strain values due to temperature increase is predicted for zigzag STs compared to SWCNTs. The results show that, for STs with radius ~5 nm, the junctions between the constituent SWCNTs play an important role in the fracture process. The Young's modulus and tensile strength were estimated for hierarchical higher-order STs using scaling laws related to the ST fractal dimension. The obtained mechanical properties suggest that STs may be used in the development of new porous, flexible, and high-strength materials.},
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, Super Carbons},
pubstate = {published},
tppubtype = {article}
}
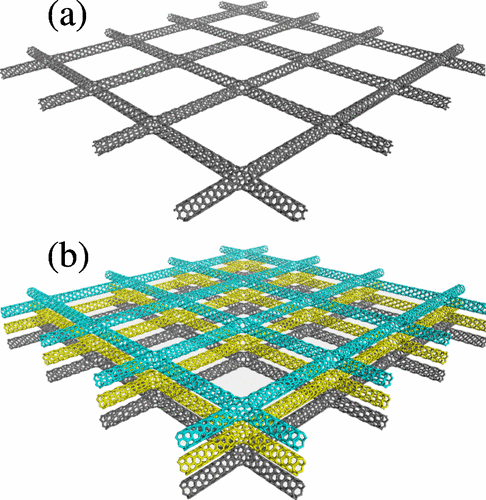
Coluci, VR; Dantas, SO; Jorio, A; Galvao, DS
Mechanical properties of carbon nanotube networks by molecular mechanics and impact molecular dynamics calculations Journal Article
In: Physical Review B, vol. 75, no. 7, pp. 075417, 2007.
Abstract | Links | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, Super Carbons
@article{coluci2007mechanical,
title = {Mechanical properties of carbon nanotube networks by molecular mechanics and impact molecular dynamics calculations},
author = {Coluci, VR and Dantas, SO and Jorio, A and Galvao, DS},
url = {http://journals.aps.org/prb/abstract/10.1103/PhysRevB.75.075417},
year = {2007},
date = {2007-01-01},
journal = {Physical Review B},
volume = {75},
number = {7},
pages = {075417},
publisher = {APS},
abstract = {We report a theoretical investigation of the mechanical properties of idealized networks formed by single-walled carbon nanotubes showing crossbar and hexagonal architectures. The study was performed by using molecular mechanics calculations and impact dynamics simulations based on bond-order empirical potential. The studied networks were predicted to have elasticity modulus of ∼10–100GPa and bulk modulus of ∼10GPa. The results show a transition from high to moderate flexibility during the deformation stages. This behavior was associated with the existence of two deformation mechanisms presented by the network related to the nanotube stretching and junction bending processes.},
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, Super Carbons},
pubstate = {published},
tppubtype = {article}
}
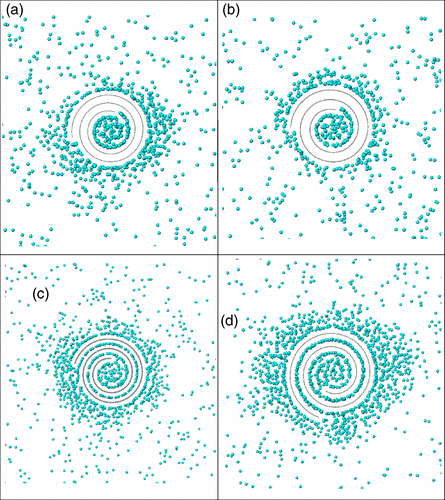
Coluci, VR; Braga, SF; Baughman, RH; Galvao, DS
Prediction of the hydrogen storage capacity of carbon nanoscrolls Journal Article
In: Physical Review B, vol. 75, no. 12, pp. 125404, 2007.
Abstract | Links | BibTeX | Tags: Hydrogen Storage, Molecular Dynamics, Monte Carlo, Scrolls
@article{coluci2007prediction,
title = {Prediction of the hydrogen storage capacity of carbon nanoscrolls},
author = {Coluci, VR and Braga, SF and Baughman, RH and Galvao, DS},
url = {http://journals.aps.org/prb/abstract/10.1103/PhysRevB.75.125404},
year = {2007},
date = {2007-01-01},
journal = {Physical Review B},
volume = {75},
number = {12},
pages = {125404},
publisher = {APS},
abstract = {Classical grand-canonical Monte Carlo simulations were performed to investigate the equilibrium hydrogen storage capacity of carbon nanoscrolls. The results show that hydrogen molecules can be absorbed in the internal cavity as well as on the external surface of the scroll when the interlayer spacing is less than 4.4Å. When the interlayer spacing is increased to 6.4Å, by assuming spacing increase due to intercalation of other species, the hydrogen molecules can also be incorporated in the interlayer galleries, doubling the gravimetric storage capacity and reaching 5.5wt% hydrogen per weight carbon at 150K and 1MPa. Our results showed that intercalated carbon nanoscrolls may be a promissing material for hydrogen storage.},
keywords = {Hydrogen Storage, Molecular Dynamics, Monte Carlo, Scrolls},
pubstate = {published},
tppubtype = {article}
}

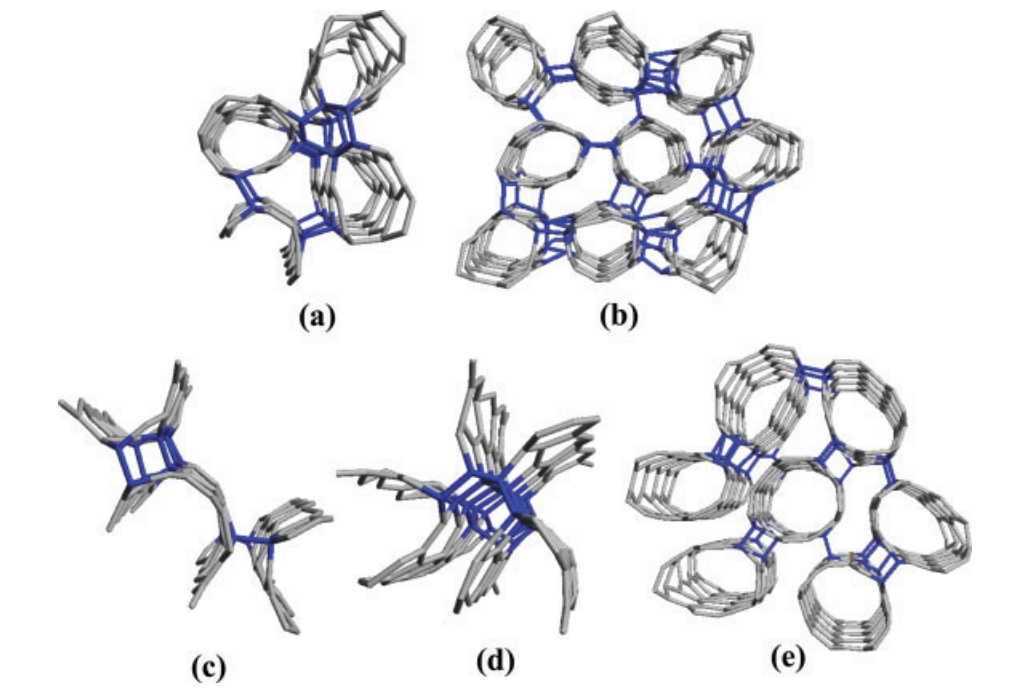
Braga, SF; Coluci, VR; Baughman, RH; Galvao, DS
Hydrogen storage in carbon nanoscrolls: An atomistic molecular dynamics study Journal Article
In: Chemical Physics Letters, vol. 441, no. 1, pp. 78–82, 2007.
Abstract | Links | BibTeX | Tags: Hydrogen Storage, Molecular Dynamics, Scrolls
@article{braga2007hydrogen,
title = {Hydrogen storage in carbon nanoscrolls: An atomistic molecular dynamics study},
author = {Braga, SF and Coluci, VR and Baughman, RH and Galvao, DS},
url = {http://www.sciencedirect.com/science/article/pii/S0009261407005209},
year = {2007},
date = {2007-01-01},
journal = {Chemical Physics Letters},
volume = {441},
number = {1},
pages = {78--82},
publisher = {North-Holland},
abstract = {We report molecular dynamics results on the hydrogen uptake in carbon nanoscrolls (CNs). CNs are formed from helically wrapped graphite layers. We observed that at low temperatures significant H2 storage is possible, but at higher temperatures thermal energies drastically reduce this capacity. Only a small fraction of hydrogen is adsorbed between scroll layers. Using temperature as the sorption/desorption variable we have observed that hydrogen can be released from the CN by temperature increase and can be readsorbed when the system is cooled. Higher capacities are expected if the CNs interlayer spacings are increased, making them an attractive nanostructure for H2 storage having fast kinetics for charge/discharge.},
keywords = {Hydrogen Storage, Molecular Dynamics, Scrolls},
pubstate = {published},
tppubtype = {article}
}
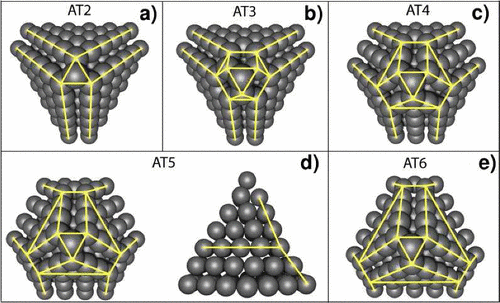
Braga, Scheila Furtado; Galvao, Douglas Soares
Molecular dynamics simulation of single wall carbon nanotubes polymerization under compression Journal Article
In: Journal of Computational Chemistry, vol. 28, no. 10, pp. 1724–1734, 2007.
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, Mechanical Properties, Molecular Dynamics, New Structures, Polymerization
@article{braga2007molecular,
title = {Molecular dynamics simulation of single wall carbon nanotubes polymerization under compression},
author = {Braga, Scheila Furtado and Galvao, Douglas Soares},
url = {http://onlinelibrary.wiley.com/store/10.1002/jcc.20684/asset/20684_ftp.pdf?v=1&t=i52l5iyb&s=94cda082eed01cd61890fffe50aad5e26cdda7d1},
year = {2007},
date = {2007-01-01},
journal = {Journal of Computational Chemistry},
volume = {28},
number = {10},
pages = {1724--1734},
publisher = {Wiley Subscription Services, Inc., A Wiley Company},
abstract = {Single wall carbon nanotubes (SWCNTs) often aggregate into bundles of hundreds of weakly interacting
tubes. Their cross-polymerization opens new possibilities for the creation of new super-hard materials. New mechanical
and electronic properties are expected from these condensed structures, as well as novel potential applications. Previous
theoretical results presented geometric modifications involving changes in the radial section of the compressed tubes
as the explanation to the experimental measurements of structural changes during tube compression. We report here
results from molecular dynamics simulations of the SWCNTs polymerization for small diameter arm chair tubes under
compression. Hydrostatic and piston-type compression of SWCNTs have been simulated for different temperatures and
rates of compression. Our results indicate that large diameter tubes (10,10) are unlike to polymerize while small diameter
ones (around 5 Å) polymerize even at room temperature. Other interesting results are the observation of the appearance
of spontaneous scroll-like structures and also the so-called tubulane motifs, which were predicted in the literature more
than a decade ago},
keywords = {Carbon Nanotubes, Mechanical Properties, Molecular Dynamics, New Structures, Polymerization},
pubstate = {published},
tppubtype = {article}
}
tubes. Their cross-polymerization opens new possibilities for the creation of new super-hard materials. New mechanical
and electronic properties are expected from these condensed structures, as well as novel potential applications. Previous
theoretical results presented geometric modifications involving changes in the radial section of the compressed tubes
as the explanation to the experimental measurements of structural changes during tube compression. We report here
results from molecular dynamics simulations of the SWCNTs polymerization for small diameter arm chair tubes under
compression. Hydrostatic and piston-type compression of SWCNTs have been simulated for different temperatures and
rates of compression. Our results indicate that large diameter tubes (10,10) are unlike to polymerize while small diameter
ones (around 5 Å) polymerize even at room temperature. Other interesting results are the observation of the appearance
of spontaneous scroll-like structures and also the so-called tubulane motifs, which were predicted in the literature more
than a decade ago
Rodrigues, V; Sato, F; Galvao, DS; Ugarte, D
Size limit of defect formation in pyramidal Pt nanocontacts Journal Article
In: Physical Review Letters, vol. 99, no. 25, pp. 255501, 2007.
Abstract | Links | BibTeX | Tags: DFT, Metallic Nanowires, Platinum, Structure, TEM, top20
@article{rodrigues2007size,
title = {Size limit of defect formation in pyramidal Pt nanocontacts},
author = {Rodrigues, V and Sato, F and Galvao, DS and Ugarte, D},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.99.255501},
year = {2007},
date = {2007-01-01},
journal = {Physical Review Letters},
volume = {99},
number = {25},
pages = {255501},
publisher = {American Physical Society},
abstract = {We report high resolution transmission electron microscopy and ab initio calculation results for defect formation in sharp pyramidal Pt nanocontacts. Our results show that there is a size limit to the existence of twins (extended structural defects). These defects are always present but blocked away from the tip axes. They may act as scattering planes, influencing the electron conductance for Pt nanocontacts at room temperature and Ag/Au nanocontacts at low temperature (<150 K).},
keywords = {DFT, Metallic Nanowires, Platinum, Structure, TEM, top20},
pubstate = {published},
tppubtype = {article}
}
Troche, KS; Coluci, VR; Rurali, R; Galvao, DS
Structural and electronic properties of zigzag carbon nanotubes filled with small fullerenes Journal Article
In: Journal of Physics: Condensed Matter, vol. 19, no. 23, pp. 236222, 2007.
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, CNT encapsulation, Electronic Structure, Fullerenes, Peapods
@article{troche2007structural,
title = {Structural and electronic properties of zigzag carbon nanotubes filled with small fullerenes},
author = {Troche, KS and Coluci, VR and Rurali, R and Galvao, DS},
url = {http://iopscience.iop.org/0953-8984/19/23/236222},
year = {2007},
date = {2007-01-01},
journal = {Journal of Physics: Condensed Matter},
volume = {19},
number = {23},
pages = {236222},
publisher = {IOP Publishing},
abstract = {In this work we investigated the encapsulation of C20 and C30 fullerenes into semiconducting carbon nanotubes to study the possibility of bandgap engineering in such systems. Classical molecular dynamics simulations coupled to tight-binding calculations were used to determine the conformational and electronic properties of carbon nanotubes with an increasing fullerene concentration. We have observed that C20 fullerenes behave similarly to a n-type dopant while C30 can provide p-type doping in some cases. The combined incorporation of both types of fullerenes (hybrid encapsulation) into the same nanotube leads to a behaviour similar to that found in electronic pn-junctions. These aspects can be exploited in the design of nanoelectronic devices using semiconducting carbon nanotubes.
},
keywords = {Carbon Nanotubes, CNT encapsulation, Electronic Structure, Fullerenes, Peapods},
pubstate = {published},
tppubtype = {article}
}
Fonseca, Alexandre F; Malta, CP; Galvao, DS
Is it possible to grow amorphous normal nanosprings? Journal Article
In: Nanotechnology, vol. 18, no. 43, pp. 435606, 2007.
Abstract | Links | BibTeX | Tags: Elasticity, Helical Structures, Mechanical Properties, Nanowires
@article{fonseca2007possible,
title = {Is it possible to grow amorphous normal nanosprings?},
author = {Fonseca, Alexandre F and Malta, CP and Galvao, DS},
url = {http://iopscience.iop.org/0957-4484/18/43/435606},
year = {2007},
date = {2007-01-01},
journal = {Nanotechnology},
volume = {18},
number = {43},
pages = {435606},
publisher = {IOP Publishing},
abstract = {Nanosprings have been objects of intense investigations in recent years. They can be classified as normal or binormal depending on the geometry of their cross-section. As normal amorphous nanosprings have not yet been observed experimentally, we have decided to investigate this matter. We discuss the shape of the catalyst in terms of the cross-sectional shape of the nanospring and show that, within the vapor–liquid–solid model, the growth of amorphous binormal nanosprings is energetically favored.},
keywords = {Elasticity, Helical Structures, Mechanical Properties, Nanowires},
pubstate = {published},
tppubtype = {article}
}
Azevedo, David L; Sato, Fernando; Galvao, Douglas S; others,
Cobaltocene encapsulation into single-walled carbon nanotubes: A molecular dynamics investigation Journal Article
In: arXiv preprint arXiv:0707.3831, 2007.
Abstract | Links | BibTeX | Tags: CNT encapsulation, Cobaltocene, Molecular Dynamics
@article{azevedo2007cobaltocene,
title = {Cobaltocene encapsulation into single-walled carbon nanotubes: A molecular dynamics investigation},
author = {Azevedo, David L and Sato, Fernando and Galvao, Douglas S and others},
url = {http://arxiv.org/abs/0707.3831},
year = {2007},
date = {2007-01-01},
journal = {arXiv preprint arXiv:0707.3831},
abstract = {Recently (PRL 96, 106804 (2006)) it was suggested that cobaltocene(CC) molecules encapsulated into (7,7) carbon nanotubes (CNT@(7,7)) could be the basis for new spintronic devices. We show here based on impact molecular dynamics and DFT calculations that when dynamical aspects are explicitly considered the CC encapsulation into CNT@(7,7) does not occur, it is prevented by a dynamic barrier mainly due to van der Waals interactions. Our results show that CNT@(13,0) having enough axial space for encapsulation but no enough one to allow freely rotation of the cobaltocene molecule would be a feasible candidate to such application.
},
keywords = {CNT encapsulation, Cobaltocene, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Rodrigues, Varlei; Sato, Fernando; Galvao, Douglas S; Ugarte, Daniel
Is Small Perfect? Size Limit to Defect Formation in Pyramidal Pt Nanocontacts Journal Article
In: arXiv preprint arXiv:0707.4187, 2007.
Abstract | Links | BibTeX | Tags: Defects, DFT, Metallic Nanowires, Structure, TEM
@article{rodrigues2007small,
title = {Is Small Perfect? Size Limit to Defect Formation in Pyramidal Pt Nanocontacts},
author = {Rodrigues, Varlei and Sato, Fernando and Galvao, Douglas S and Ugarte, Daniel},
url = {http://xxx.tau.ac.il/abs/0707.4187},
year = {2007},
date = {2007-01-01},
journal = {arXiv preprint arXiv:0707.4187},
abstract = {We report high resolution transmission electron microscopy and ab initio calculation results for the defect formation in Pt nanocontacts (NCs). Our results show that there is a size limit to the existence of twins (extended structural defects). Defects are always present but blocked away from the tip axes. The twins may act as scattering plane, influencing contact electron transmission for Pt NC at room temperature and Ag/Au NC at low temperature.},
keywords = {Defects, DFT, Metallic Nanowires, Structure, TEM},
pubstate = {published},
tppubtype = {article}
}
Troche, Karla S; Coluci, Vitor R; Galvao, Douglas S
Atomistic study of the encapsulation of diamondoids inside carbon nanotubes Journal Article
In: arXiv preprint arXiv:0707.1777, 2007.
Abstract | Links | BibTeX | Tags: CNT encapsulation, Diamondoids, Molecular Dynamics
@article{troche2007atomistic,
title = {Atomistic study of the encapsulation of diamondoids inside carbon nanotubes},
author = {Troche, Karla S and Coluci, Vitor R and Galvao, Douglas S},
url = {http://arxiv.org/abs/0707.1777},
year = {2007},
date = {2007-01-01},
journal = {arXiv preprint arXiv:0707.1777},
abstract = {The encapsulation of hydrogen-terminated nanosized diamond fragments (the so-called diamondoids) into armchair single walled carbon nanotubes with diameters in the range of 1.0 up to 2.2 nm has been investigated using classical molecular dynamics simulations. Diameter dependent molecular ordered phases were found for the encapsulation of adamantane (C10H16), diamantane (C14H20), and dihydroxy diamantane (C14H20O2). The same types of chiral ordered phases (double, triple, 4- and 5-stranded helices) observed for the encapsulation of C60 molecules were also observed for diamondoids. On the other hand, some achiral phases comprising layered structures were not observed. Our results also indicate that the modification of diamantane through functionalization with hydroxyl groups can lead to an enhancement of the packing of molecules inside the nanotubes compared to nonfunctionalized compounds. Comparisons to hard-sphere models are also presented revealing differences, specially when more asymmetrical diamondoids are considered. For larger structures (adamantane tetramers) we have not observed long-range ordering for nanotubes with diameters in the range of 1.49 to 2.17 nm but only a tendency to form incomplete helical structures.},
keywords = {CNT encapsulation, Diamondoids, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Sato, F; Legoas, SB; Otero, R; Hummelink, F; Thostrup, P; Lægsgaard, E; Stensgaard, I; Besenbacher, F; Galvao, DS
Molecular Recognition Effects in the Surface Diffusion of Large Organic Molecules: The Case of Violet Lander Journal Article
In: arXiv preprint arXiv:0708.2915, 2007.
Abstract | Links | BibTeX | Tags: Molecular Dynamics, Molecular Machines, Organic-Inorganic Interfaces, Violet Landers
@article{sato2007molecular,
title = {Molecular Recognition Effects in the Surface Diffusion of Large Organic Molecules: The Case of Violet Lander},
author = {Sato, F and Legoas, SB and Otero, R and Hummelink, F and Thostrup, P and Lægsgaard, E and Stensgaard, I and Besenbacher, F and Galvao, DS},
url = {http://xxx.tau.ac.il/pdf/0708.2915.pdf},
year = {2007},
date = {2007-01-01},
journal = {arXiv preprint arXiv:0708.2915},
abstract = {Violet Lander (VL) (C108H104) is a large organic molecule that when deposited on Cu (110)
exhibited lock-and-key like behavior (Otero et al., Nature Mater. 3, 779 (2004)). In this work we
report on a detailed fully atomistic molecular dynamics study of this phenomenon. Our results show
that it has its physical basis in the interplay of the molecular hydrogens and the Cu(110) atomic
spacing, which is a direct consequence of an accidental commensurability between molecule and
surface dimensions. This knowledge could be used to engineer new molecules capable of displaying
lock-and-key behavior with new potential applications in nanotechology},
keywords = {Molecular Dynamics, Molecular Machines, Organic-Inorganic Interfaces, Violet Landers},
pubstate = {published},
tppubtype = {article}
}
exhibited lock-and-key like behavior (Otero et al., Nature Mater. 3, 779 (2004)). In this work we
report on a detailed fully atomistic molecular dynamics study of this phenomenon. Our results show
that it has its physical basis in the interplay of the molecular hydrogens and the Cu(110) atomic
spacing, which is a direct consequence of an accidental commensurability between molecule and
surface dimensions. This knowledge could be used to engineer new molecules capable of displaying
lock-and-key behavior with new potential applications in nanotechology
Sato, Fernando; Braga, Scheila F; Santos, Helio F dos; Galvao, Douglas S
Structure-Activity Relationship Investigation of Some New Tetracyclines by Electronic Index Methodology Journal Article
In: arXiv preprint arXiv:0708.2931, 2007.
Abstract | Links | BibTeX | Tags: Drug Design, Electronic Structure, Neural Networks, PCA/HCA, Tetracyclines, Theory of Electronic Indices
@article{sato2007structure,
title = {Structure-Activity Relationship Investigation of Some New Tetracyclines by Electronic Index Methodology},
author = {Sato, Fernando and Braga, Scheila F and Santos, Helio F dos and Galvao, Douglas S},
url = {http://arxiv.org/abs/0708.2931},
year = {2007},
date = {2007-01-01},
journal = {arXiv preprint arXiv:0708.2931},
abstract = {Tetracyclines are an old class of molecules that constitute a broad-spectrum antibiotics. Since the first member of tetracycline family were isolated, the clinical importance of these compounds as therapeutic and prophylactic agents against a wide range of infections has stimulated efforts to define their mode of action as inhibitors of bacterial reproduction. We used three SAR methodologies for the analysis of biological activity of a set of 104 tetracycline compounds. Our calculation were carried out using the semi-empirical Austin Method One (AM1) and Parametric Method 3 (PM3). Electronic Indices Methodology (EIM), Principal Component Analysis (PCA) and Artificial Neural Networks (ANN) were applied to the classification of 14 old and 90 new proposed derivatives of tetracyclines. Our results make evident the importance of EIM descriptors in pattern recognition and also show that the EIM can be effectively used to predict the biological activity of Tetracyclines.},
keywords = {Drug Design, Electronic Structure, Neural Networks, PCA/HCA, Tetracyclines, Theory of Electronic Indices},
pubstate = {published},
tppubtype = {article}
}
Coluci, VR; Dantas, SO; Jorio, A; Galvao, DS
Electronic and Mechanical Properties of Super Carbon Nanotube Networks Proceedings
Warrendale, Pa.; Materials Research Society; 1999, vol. 963, 2007.
Abstract | Links | BibTeX | Tags: Fracture, Mechanical Properties, Molecular Dynamics, Super Carbons
@proceedings{coluci2007electronic,
title = {Electronic and Mechanical Properties of Super Carbon Nanotube Networks},
author = {Coluci, VR and Dantas, SO and Jorio, A and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8026810&fulltextType=RA&fileId=S1946427400054014},
year = {2007},
date = {2007-01-01},
journal = {MATERIALS RESEARCH SOCIETY SYMPOSIUM PROCEEDINGS},
volume = {963},
pages = {1},
publisher = {Warrendale, Pa.; Materials Research Society; 1999},
abstract = {Eletronic and mechanical properties of ordered carbon nanotube networks are studied using molecular dynamics simulations and tight-binding calculations. These networks are formed by single walled carbon nanotubes (SWNT) regularly connected by junctions. The use of different types of junctions (“Y”-, “X”-like junctions, for example) allows the construction of networks with different symmetries. These networks can be very flexible and the elastic deformation was associated with two main deformation mechanisms (bending and stretching ) of the constituents SWNTs. Rolling up the networks, “super” carbon nanotubes can be constructed. These super-tubes share some of the main electronic features of the SWNT which form them but important changes are predicted (e.g. reduction of bandgap value). Simulations of their deformations under tensile stress have revealed that the super-tubes are softer than the corresponding SWNT and that their rupture occur in higher strain values.},
keywords = {Fracture, Mechanical Properties, Molecular Dynamics, Super Carbons},
pubstate = {published},
tppubtype = {proceedings}
}

Pugno, Nicola; Coluci, V; Galvao, DS
Nanotube-or graphene-based nanoarmors Book Chapter
In: Computational & Experimental Analysis of Damaged Materials, pp. 145-154 , 2007.
Abstract | Links | BibTeX | Tags: Elasticity, Mechanical Properties, Molecular Dynamics, Super Carbons
@inbook{pugno2007nanotube,
title = {Nanotube-or graphene-based nanoarmors},
author = {Pugno, Nicola and Coluci, V and Galvao, DS},
url = {http://www.ing.unitn.it/~pugno/NP_PDF/IV/5-COLUCI07.pdf},
year = {2007},
date = {2007-01-01},
booktitle = {Computational & Experimental Analysis of Damaged Materials},
pages = {145-154 },
abstract = { In this paper, nanoimpacts on hexagonal or
crossbar nanotube networks as well as on graphene
sheets are investigated by elasticity and finite
kinematics or impact molecular dynamic simulations.
A transition from bending to stretching by increasing
the impact kinetic energy of the nanoprojectile is
clearly observed. The analysis suggests that the
investigated nanotextures are ideal for designing
futuristic nanoarmors. },
keywords = {Elasticity, Mechanical Properties, Molecular Dynamics, Super Carbons},
pubstate = {published},
tppubtype = {inbook}
}
crossbar nanotube networks as well as on graphene
sheets are investigated by elasticity and finite
kinematics or impact molecular dynamic simulations.
A transition from bending to stretching by increasing
the impact kinetic energy of the nanoprojectile is
clearly observed. The analysis suggests that the
investigated nanotextures are ideal for designing
futuristic nanoarmors.

Fonseca, AD; Malta, CP; Galvao, DS
Elastic Properties of Normal and Binormal Helical Nanowires Proceedings
Warrendale, Pa.; Materials Research Society; 1999, vol. 963, 2007.
Abstract | Links | BibTeX | Tags: Elasticity, Helical Structures, Mechanical Properties, Nanowires
@proceedings{fonseca2007elastic,
title = {Elastic Properties of Normal and Binormal Helical Nanowires},
author = {Fonseca, AD and Malta, CP and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8026852},
year = {2007},
date = {2007-01-01},
journal = {MATERIALS RESEARCH SOCIETY SYMPOSIUM PROCEEDINGS},
volume = {963},
pages = {88},
publisher = {Warrendale, Pa.; Materials Research Society; 1999},
abstract = {A helical nanowire can be defined as being a nanoscopic rod whose axis follows a helical curve in space. In the case of a nanowire with asymmetric cross section, the helical nanostructure can be classified as normal or binormal helix, according to the orientation of the cross section with respect to the helical axis of the structure. In this work, we present a simple model to study the elastic properties of a helical nanowire with asymmetric cross section. We use the framework of the Kirchhoff rod model to obtain an expression relating the Hooke's constant, h, of normal and binormal nanohelices to their geometric features. We also obtain the Young's modulus values. These relations can be used by experimentalists to evaluate the elastic properties of helical nanostructures. We showed that the Hooke's constant of a normal nanohelix is higher than that of a binormal one. We illustrate our results using experimentally obtained nanohelices reported in the literature.},
keywords = {Elasticity, Helical Structures, Mechanical Properties, Nanowires},
pubstate = {published},
tppubtype = {proceedings}
}
2006
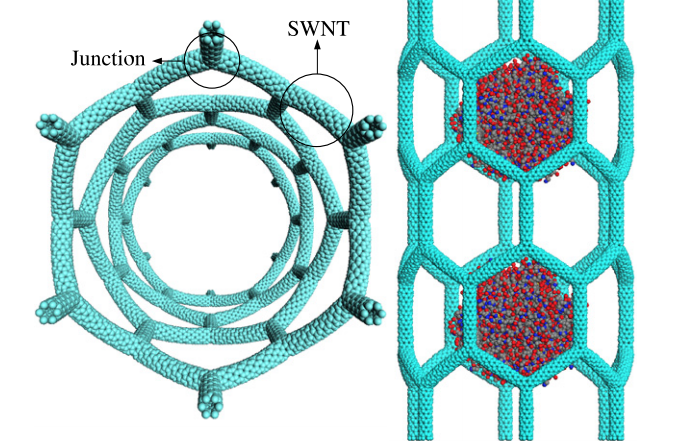
Coluci, Vitor R; Galvao, Douglas S; Jorio, A
Geometric and electronic structure of carbon nanotube networks:'super'-carbon nanotubes Journal Article
In: Nanotechnology, vol. 17, no. 3, pp. 617, 2006.
Abstract | Links | BibTeX | Tags: DFT, Mechanical Properties, Molecular Dynamics, Super Carbons
@article{coluci2006geometric,
title = {Geometric and electronic structure of carbon nanotube networks:'super'-carbon nanotubes},
author = {Coluci, Vitor R and Galvao, Douglas S and Jorio, A},
url = {http://iopscience.iop.org/0957-4484/17/3/001},
year = {2006},
date = {2006-01-01},
journal = {Nanotechnology},
volume = {17},
number = {3},
pages = {617},
publisher = {IoP Publishing},
abstract = {Structures of the so-called super-carbon nanotubes are proposed. These structures are built from single walled carbon nanotubes connected by Y-like junctions forming a 'super'-sheet that is then rolled into a seamless cylinder. Such a procedure can be repeated several times, generating a fractal structure. This procedure is not limited to carbon nanotubes, and can be easily modified for application to other systems. Tight binding total energy and density of states calculations showed that the 'super'-sheets and tubes are stable and predicted to present metallic and semiconducting behaviour.},
keywords = {DFT, Mechanical Properties, Molecular Dynamics, Super Carbons},
pubstate = {published},
tppubtype = {article}
}
Bettini, Jefferson; Sato, Fernando; Coura, Pablo Zimmerman; Dantas, SO; Galvao, Douglas Soares; Ugarte, Daniel
Experimental realization of suspended atomic chains composed of different atomic species Journal Article
In: Nature Nanotechnology, vol. 1, no. 3, pp. 182–185, 2006.
Abstract | Links | BibTeX | Tags: Metallic Nanowires, Molecular Dynamics, TEM, top20
@article{bettini2006experimental,
title = {Experimental realization of suspended atomic chains composed of different atomic species},
author = {Bettini, Jefferson and Sato, Fernando and Coura, Pablo Zimmerman and Dantas, SO and Galvao, Douglas Soares and Ugarte, Daniel},
url = {http://www.nature.com/nnano/journal/v1/n3/full/nnano.2006.132.html},
year = {2006},
date = {2006-01-01},
journal = {Nature Nanotechnology},
volume = {1},
number = {3},
pages = {182--185},
publisher = {Nature Publishing Group},
abstract = {Research into nanostructured materials frequently relates to pure substances. This contrasts with industrial applications, where chemical doping or alloying is often used to enhance the electrical or mechanical properties of materials1. However, the controlled preparation of doped nanomaterials has been much more difficult than expected because the increased surface-area-to-volume ratio can, for instance, lead to the expulsion of impurities (self-purification)2. For nanostructured alloys, the influence of growth methods and the atomic structure on self-purification is still open to investigation2, 3. Here, we explore, experimentally and with molecular dynamics simulations, to what extent alloying persists in the limit that a binary metal is mechanically stretched to a linear chain of atoms. Our results reveal a gradual evolution of the arrangement of the different atomic elements in the narrowest region of the chain, where impurities may be expelled to the surface or enclosed during elongation.
},
keywords = {Metallic Nanowires, Molecular Dynamics, TEM, top20},
pubstate = {published},
tppubtype = {article}
}
Sato, F; Moreira, AS; Bettini, J; Coura, PZ; Dantas, SO; Ugarte, D; Galvao, DS
Transmission electron microscopy and molecular dynamics study of the formation of suspended copper linear atomic chains Journal Article
In: Physical Review-Section B-Condensed Matter, vol. 74, no. 19, pp. 193401–193401, 2006.
Abstract | Links | BibTeX | Tags: Linear Atomic Chains, Metallic Nanowires, Molecular Dynamics, TEM
@article{sato2006surface,
title = {Transmission electron microscopy and molecular dynamics study of the formation of suspended copper linear atomic chains},
author = {Sato, F and Moreira, AS and Bettini, J and Coura, PZ and Dantas, SO and Ugarte, D and Galvao, DS},
url = {http://journals.aps.org/prb/abstract/10.1103/PhysRevB.74.193401},
year = {2006},
date = {2006-01-01},
journal = {Physical Review-Section B-Condensed Matter},
volume = {74},
number = {19},
pages = {193401--193401},
publisher = {Woodbury, NY: published by the American Physical Society through the American Institute of Physics, c1998-},
abstract = {We report high-resolution transmission electron microscopy and molecular dynamics simulation results of mechanically stretching nanowires leading to linear atomic suspended chain (LAC) formation. In contrast with some previous experimental and theoretical works in the literature that stated that the formation of LAC’s for copper should be unlikely our results showed the existence of LAC’s for the [111], [110], and [100] crystallographic directions, being thus the sequence of most probable occurrence. Our results clearly indicate that temperture and pulling velocity, associated with internal stress, are fundamental aspects to determine LAC formation.},
keywords = {Linear Atomic Chains, Metallic Nanowires, Molecular Dynamics, TEM},
pubstate = {published},
tppubtype = {article}
}
Lorite, Gabriela S; Coluci, Vitor R; da Silva, Maria Ivonete N; Deziderio, Shirlei N; Graeff, Carlos Frederico O; Galvao, Douglas S; Cotta, Monica A
Synthetic melanin films: Assembling mechanisms, scaling behavior, and structural properties Journal Article
In: Journal of Applied Physics, vol. 99, no. 11, pp. 113511, 2006.
Abstract | Links | BibTeX | Tags: Electronic Structure, Melanin, Structure
@article{lorite2006synthetic,
title = {Synthetic melanin films: Assembling mechanisms, scaling behavior, and structural properties},
author = {Lorite, Gabriela S and Coluci, Vitor R and da Silva, Maria Ivonete N and Deziderio, Shirlei N and Graeff, Carlos Frederico O and Galvao, Douglas S and Cotta, Monica A},
url = {http://scitation.aip.org/content/aip/journal/jap/99/11/10.1063/1.2200401},
year = {2006},
date = {2006-01-01},
journal = {Journal of Applied Physics},
volume = {99},
number = {11},
pages = {113511},
publisher = {AIP Publishing},
abstract = {In this work we report on the surface characterization of melanin thin films prepared using both water-based and organic solvent-based melanin syntheses. Atomic force microscopy(AFM) analysis of these films suggests that the organic solvent synthesis provides relatively planar basic melanin structures; these basic structures generate surface steps with height in the range of 2–3nm and small tendency to form larger aggregates. The scaling properties obtained from the AFM data were used to infer the assembling mechanisms of these thin films which depend on the solvent used for melanin synthesis. The behavior observed in organic solvent-based melanin suggests a diffusion-limited aggregation process. Thus films with good adhesion to the substrate and smoother morphologies than water-prepared melanin films are obtained. Electronic structure calculations using a conductorlike screening model were also performed in order to elucidate the microscopic processes of thin film formation. Our results suggest that the agglomerates observed in hydrated samples originate from reaction with water at specific locations on the surface most likely defects on the planar structure.},
keywords = {Electronic Structure, Melanin, Structure},
pubstate = {published},
tppubtype = {article}
}
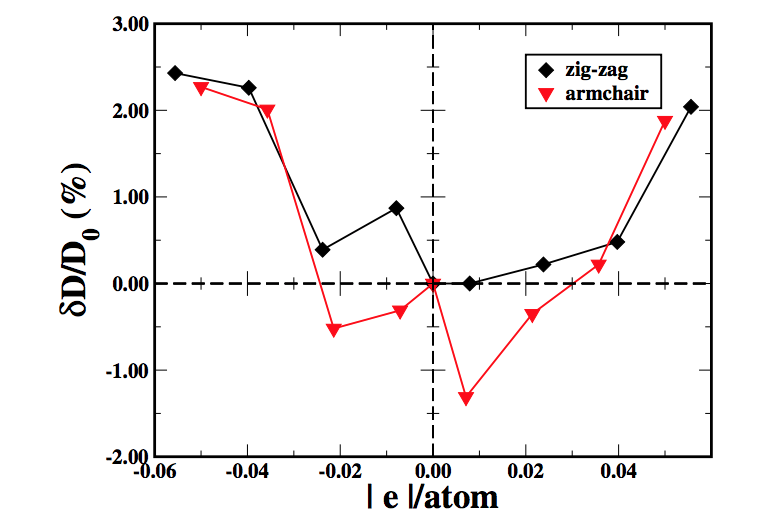
Rurali, R; Coluci, VR; Galvao, DS
Prediction of giant electroactuation for papyruslike carbon nanoscroll structures: first-principles calculations Journal Article
In: Physical Review B, vol. 74, no. 8, pp. 085414, 2006.
Abstract | Links | BibTeX | Tags: DFT, Electronic Structure, Eletroactuation, Scrolls
@article{rurali2006prediction,
title = {Prediction of giant electroactuation for papyruslike carbon nanoscroll structures: first-principles calculations},
author = {Rurali, R and Coluci, VR and Galvao, DS},
url = {http://journals.aps.org/prb/abstract/10.1103/PhysRevB.74.085414},
year = {2006},
date = {2006-01-01},
journal = {Physical Review B},
volume = {74},
number = {8},
pages = {085414},
publisher = {American Physical Society},
abstract = {We study by first-principles calculations the electromechanical response of carbon nanoscroll structures. We show that although they present a very similar behavior to carbon nanotubes in their axial deformation sensitivity, they exhibit a radial response upon charge injection which is up to one order of magnitude larger. In association with their high stability, this behavior makes them a natural choice for a new class of very efficient nanoactuators.},
keywords = {DFT, Electronic Structure, Eletroactuation, Scrolls},
pubstate = {published},
tppubtype = {article}
}
Braga, SF; Galvao, DS
Single wall carbon nanotubes polymerization under compression: An atomistic molecular dynamics study Journal Article
In: Chemical physics letters, vol. 419, no. 4, pp. 394–399, 2006.
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, Molecular Dynamics, Polymerization
@article{braga2006single,
title = {Single wall carbon nanotubes polymerization under compression: An atomistic molecular dynamics study},
author = {Braga, SF and Galvao, DS},
url = {http://www.sciencedirect.com/science/article/pii/S0009261405018592},
year = {2006},
date = {2006-01-01},
journal = {Chemical physics letters},
volume = {419},
number = {4},
pages = {394--399},
publisher = {Elsevier},
abstract = {Recently, it was reported experimental observations of crosslinking between carbon nanotubes (CNTs) under pressure. Similarly to CNT growth formation the details of these polymerization processes are still unclear. In this work, we report a molecular dynamics simulation of the polymerization of a bundle of single-wall carbon nanotubes under compression using Brenner reactive potentials. Our results show that for small tube diameters extensive crosslinking formation can occur. For larger tube diameter, we obtained the first theoretical evidences that scroll-like structures (recently experimentally obtained) can be formed from SWCNTs.
},
keywords = {Carbon Nanotubes, Molecular Dynamics, Polymerization},
pubstate = {published},
tppubtype = {article}
}
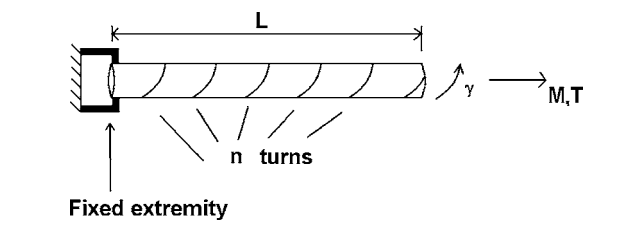
da Fonseca, Alexandre F; Malta, CP; Galvao, Douglas S
Elastic properties of nanowires Journal Article
In: Journal of Applied Physics, vol. 99, no. 9, pp. 094310, 2006.
Abstract | Links | BibTeX | Tags: Elasticity, Filaments, Nanowires
@article{da2006elastic,
title = {Elastic properties of nanowires},
author = {da Fonseca, Alexandre F and Malta, CP and Galvao, Douglas S},
url = {http://scitation.aip.org/content/aip/journal/jap/99/9/10.1063/1.2194309},
year = {2006},
date = {2006-01-01},
journal = {Journal of Applied Physics},
volume = {99},
number = {9},
pages = {094310},
publisher = {AIP Publishing},
abstract = {We present a model to study Young’s modulus and Poisson’s ratio of the composite material of amorphous nanowires. It is an extension of the model derived by two of us [da Fonseca and Galvão, Phys. Rev. Lett.92, 175502 (2004)] to study the elastic properties of amorphous nanosprings. The model is based on twisting and tensioning a straight nanowire and we propose an experimental setup to obtain the elastic parameters of the nanowire. We used the Kirchhoff rod model to obtain the expressions for the elastic constants of the nanowire.
},
keywords = {Elasticity, Filaments, Nanowires},
pubstate = {published},
tppubtype = {article}
}
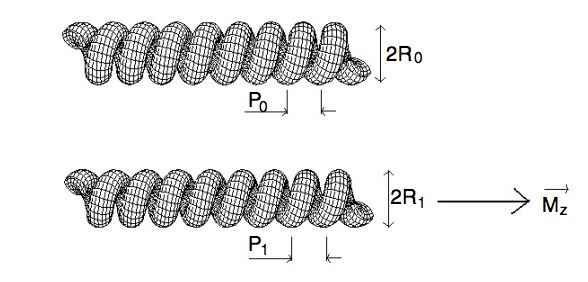
da Fonseca, Alexandre F; Malta, CP; Galvao, DS
Mechanical properties of amorphous nanosprings Journal Article
In: Nanotechnology, vol. 17, no. 22, pp. 5620, 2006.
Abstract | Links | BibTeX | Tags: Elasticity, Filaments, Nanowires
@article{da2006mechanical,
title = {Mechanical properties of amorphous nanosprings},
author = {da Fonseca, Alexandre F and Malta, CP and Galvao, DS},
url = {http://iopscience.iop.org/0957-4484/17/22/015},
year = {2006},
date = {2006-01-01},
journal = {Nanotechnology},
volume = {17},
number = {22},
pages = {5620},
publisher = {IOP Publishing},
abstract = {Helical amorphous nanosprings have attracted particular interest due to their special mechanical properties. In this work we present a simple model, within the framework of the Kirchhoff rod model, for investigating the structural properties of nanosprings having asymmetric cross section. We have derived expressions that can be used to obtain the Young's modulus and Poisson's ratio of the nanospring material composite. We also address the importance of the presence of a catalyst in the growth process of amorphous nanosprings in terms of the stability of helical rods.
},
keywords = {Elasticity, Filaments, Nanowires},
pubstate = {published},
tppubtype = {article}
}
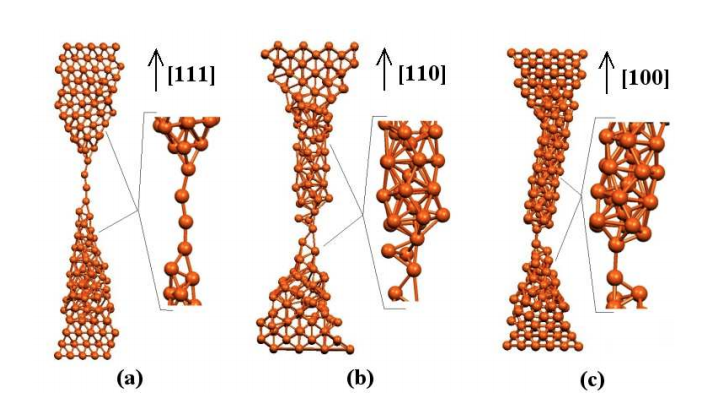
Sato, F; Moreira, AS; Bettini, J; Coura, PZ; Dantas, SO; Ugarte, D; Galvao, DS
On the Formation of Copper Linear Atomic Suspended Chains Journal Article
In: arXiv preprint cond-mat/0602092, 2006.
Abstract | Links | BibTeX | Tags: Copper, Linear Atomic Chains, Metallic Nanowires, Molecular Dynamics, TEM
@article{sato2006formation,
title = {On the Formation of Copper Linear Atomic Suspended Chains},
author = {Sato, F and Moreira, AS and Bettini, J and Coura, PZ and Dantas, SO and Ugarte, D and Galvao, DS},
url = {http://arxiv.org/abs/cond-mat/0602092},
year = {2006},
date = {2006-01-01},
journal = {arXiv preprint cond-mat/0602092},
abstract = {We report high resolution transmission electron microscopy and classical molecular dynamics simulation results of mechanically stretching copper nanowires conducting to linear atomic suspended chains (LACs) formation. In contrast with some previous experimental and theoretical work in literature that stated that the formation of LACs for copper should not exist our results showed the existence of LAC for the [111], [110], and [100] crystallographic directions, being thus the sequence of most probable occurence.},
keywords = {Copper, Linear Atomic Chains, Metallic Nanowires, Molecular Dynamics, TEM},
pubstate = {published},
tppubtype = {article}
}
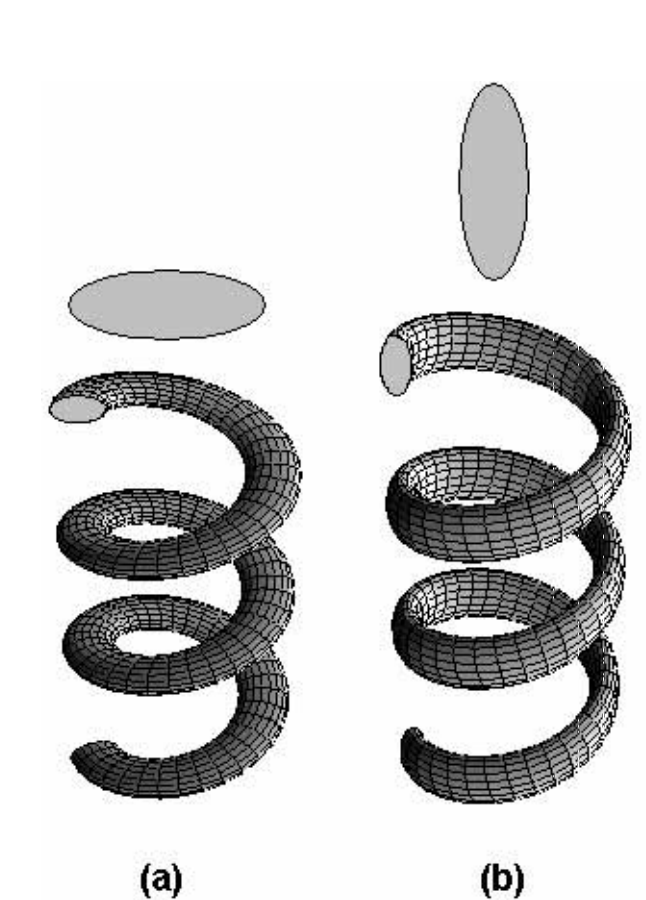
da Fonseca, Alexandre Fontes; Malta, CP; Galvao, Douglas S
Elastic Properties of Normal and Binormal Helical Nanowires Proceedings
Cambridge University Press, vol. 963, 2006.
Abstract | Links | BibTeX | Tags: Elasticity, Filaments, Metallic Nanowires
@proceedings{da2006elasticb,
title = {Elastic Properties of Normal and Binormal Helical Nanowires},
author = {da Fonseca, Alexandre Fontes and Malta, CP and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8026852},
year = {2006},
date = {2006-01-01},
journal = {MRS Proceedings},
volume = {963},
pages = {0963--Q20},
publisher = {Cambridge University Press},
abstract = {A helical nanowire can be defined as being a nanoscopic rod whose axis follows a helical curve in space. In the case of a nanowire with asymmetric cross section, the helical nanostructure can be classified as normal or binormal helix, according to the orientation of the cross section with respect to the helical axis of the structure. In this work, we present a simple model to study the elastic properties of a helical nanowire with asymmetric cross section. We use the framework of the Kirchhoff rod model to obtain an expression relating the Hooke's constant, h, of normal and binormal nanohelices to their geometric features. We also obtain the Young's modulus values. These relations can be used by experimentalists to evaluate the elastic properties of helical nanostructures. We showed that the Hooke's constant of a normal nanohelix is higher than that of a binormal one. We illustrate our results using experimentally obtained nanohelices reported in the literature.},
keywords = {Elasticity, Filaments, Metallic Nanowires},
pubstate = {published},
tppubtype = {proceedings}
}
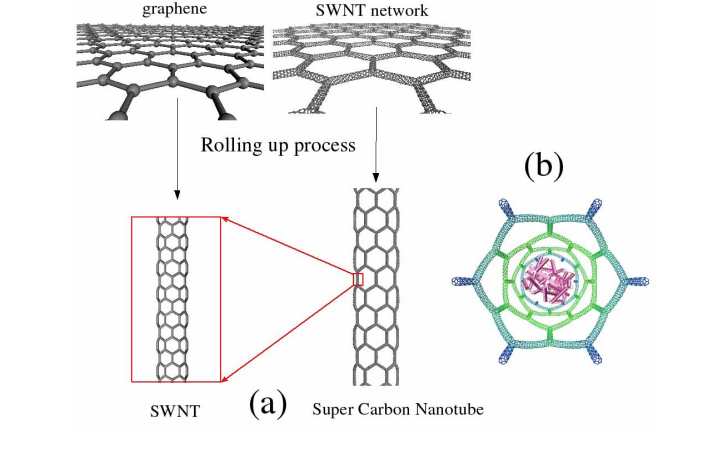
Coluci, Vitor R; Dantas, Socrates O; Jorio, Ado; Galvao, Douglas S.
Electronic and Mechanical Properties of Super Carbon Nanotube Networks Proceedings
Cambridge University Press, vol. 963, 2006.
Abstract | Links | BibTeX | Tags: Mechanical Properties, Molecular Dynamics, Super Carbons
@proceedings{coluci2006electronic,
title = {Electronic and Mechanical Properties of Super Carbon Nanotube Networks},
author = {Coluci, Vitor R and Dantas, Socrates O and Jorio, Ado and Galvao, Douglas S.},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8026810&fileId=S1946427400054014},
year = {2006},
date = {2006-01-01},
journal = {MRS Proceedings},
volume = {963},
pages = {0963--Q15},
publisher = {Cambridge University Press},
abstract = {Eletronic and mechanical properties of ordered carbon nanotube networks are studied using molecular dynamics simulations and tight-binding calculations. These networks are formed by single walled carbon nanotubes (SWNT) regularly connected by junctions. The use of different types of junctions (“Y”-, “X”-like junctions, for example) allows the construction of networks with different symmetries. These networks can be very flexible and the elastic deformation was associated with two main deformation mechanisms (bending and stretching ) of the constituents SWNTs. Rolling up the networks, “super” carbon nanotubes can be constructed. These super-tubes share some of the main electronic features of the SWNT which form them but important changes are predicted (e.g. reduction of bandgap value). Simulations of their deformations under tensile stress have revealed that the super-tubes are softer than the corresponding SWNT and that their rupture occur in higher strain values.},
keywords = {Mechanical Properties, Molecular Dynamics, Super Carbons},
pubstate = {published},
tppubtype = {proceedings}
}
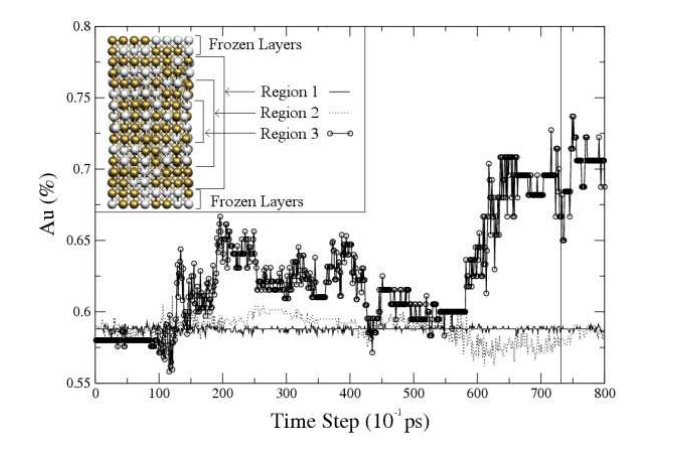
Bettini, J; Sato, F; Coura, PZ; Dantas, SO; Galvao, DS; Ugarte, D
Nanowires and Suspended Atom Chains from Metal alloys Journal Article
In: arXiv preprint cond-mat/0601617, 2006.
Abstract | Links | BibTeX | Tags: Molecular Dynamics, TEM
@article{bettini2006nanowires,
title = {Nanowires and Suspended Atom Chains from Metal alloys},
author = {Bettini, J and Sato, F and Coura, PZ and Dantas, SO and Galvao, DS and Ugarte, D},
url = {http://arxiv.org/abs/cond-mat/0601617},
year = {2006},
date = {2006-01-01},
journal = {arXiv preprint cond-mat/0601617},
abstract = {We present a study of the elongation and rupture of gold-silver alloy nanowires. Atomistic details of the evolution were derived from time-resolved atomic resolution transmission electron microscopy and molecular dynamics simulations. The results show the occurrence of gold enrichment at the nanojunction region, leading to a gold-like structural behavior even for alloys with minor gold content. Our observations have also revealed the formation of mixed (Au and Ag) linear atomic chains.},
keywords = {Molecular Dynamics, TEM},
pubstate = {published},
tppubtype = {article}
}

Rurali, R; Coluci, VR; Galvao, DS
Prediction of Giant Electro-actuation for Carbon Nanoscrolls Journal Article
In: arXiv preprint cond-mat/0603239, 2006.
Abstract | Links | BibTeX | Tags: DFT, Electroactuation, Electronic Structure, Scrolls
@article{rurali2006predictionb,
title = {Prediction of Giant Electro-actuation for Carbon Nanoscrolls},
author = {Rurali, R and Coluci, VR and Galvao, DS},
url = {http://arxiv.org/abs/cond-mat/0603239},
year = {2006},
date = {2006-01-01},
journal = {arXiv preprint cond-mat/0603239},
abstract = {We study by first-principles calculations the electro-mechanical response of carbon nanoscrolls. We show that although they present a very similar behavior to carbon nanotubes for what concerns the axial deformation sensitivity, they exhibit a radial response upon charge injection which is up to one order of magnitude larger. In association with their high stability, this behavior make them a natural choice for a new class of very efficient nano-actuators.},
keywords = {DFT, Electroactuation, Electronic Structure, Scrolls},
pubstate = {published},
tppubtype = {article}
}
Troche, KS; Coluci, VR; Rurali, R; Galvao, DS
Doping of zigzag carbon nanotubes through the encapsulation of small fullerenes Journal Article
In: arXiv preprint cond-mat/0607197, 2006.
Abstract | Links | BibTeX | Tags: CNT encapsulation, DFT, Molecular Dynamics
@article{troche2006doping,
title = {Doping of zigzag carbon nanotubes through the encapsulation of small fullerenes},
author = {Troche, KS and Coluci, VR and Rurali, R and Galvao, DS},
url = {http://arxiv.org/abs/cond-mat/0607197},
year = {2006},
date = {2006-01-01},
journal = {arXiv preprint cond-mat/0607197},
abstract = {In this work we investigated the encapsulation of C20 and C30 fullerenes into semiconducting carbon nanotubes to study the possibility of bandgap engineering in such systems. Classical molecular dynamics simulations coupled to tight-binding calculations were used to determine the conformational and electronic properties of carbon nanotube supercells containing up to 12 fullerenes. We have observed that C20 fullerenes behave similarly to a p-type dopant while C30 ones work as n-type ones. For larger diameter nanotubes, where fullerene patterns start to differ from the linear arrangements (peapods), the doping features are preserved for both fullerenes, but local disorder plays an important role and significantly alters the electronic structure. The combined incorporation of both fullerene types (hybrid encapsulation) into the same nanotube leads to a behavior similar to that found in electronic junctions in Silicon-based electronic devices. These aspects can be exploited in the design of nanoelectronic devices using semiconducting carbon nanotubes.},
keywords = {CNT encapsulation, DFT, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
2005
Troche, Karla S; Coluci, Vitor R; Braga, Scheila F; Chinellato, David D; Sato, Fernando; Legoas, Sergio B; Rurali, Riccardo; Galvao, Douglas S
Prediction of ordered phases of encapsulated C60, C70, and C78 inside carbon nanotubes Journal Article
In: Nano letters, vol. 5, no. 2, pp. 349–355, 2005.
Abstract | Links | BibTeX | Tags: Molecular Dynamics, Scrolls
@article{troche2005prediction,
title = {Prediction of ordered phases of encapsulated C60, C70, and C78 inside carbon nanotubes},
author = {Troche, Karla S and Coluci, Vitor R and Braga, Scheila F and Chinellato, David D and Sato, Fernando and Legoas, Sergio B and Rurali, Riccardo and Galvao, Douglas S},
url = {http://pubs.acs.org/doi/abs/10.1021/nl047930r},
year = {2005},
date = {2005-01-01},
journal = {Nano letters},
volume = {5},
number = {2},
pages = {349--355},
publisher = {ACS Publications},
abstract = {arbon nanotube scrolls (CNSs) provide an interesting form of carbon that ideally consists of a single sheet of graphite that is spiral wrapped to form a nanotube. We here use molecular dynamics simulations to investigate CNS formation, stability, and the structural effects due to charge injection. CNS formation is seen to automatically occur when a critical overlap between sheet layers is achieved for the partially curled sheet. We find that charge injection causes unwinding of the CNSs, which might be important for the application of CNSs as nanomechanical
actuators.
},
keywords = {Molecular Dynamics, Scrolls},
pubstate = {published},
tppubtype = {article}
}
actuators.
Coluci, VR; Legoas, SB; de Aguiar, MAM; Galvao, DS
Chaotic signature in the motion of coupled carbon nanotube oscillators Journal Article
In: Nanotechnology, vol. 16, no. 4, pp. 583, 2005.
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, Chaos, Oscillators
@article{coluci2005chaotic,
title = {Chaotic signature in the motion of coupled carbon nanotube oscillators},
author = {Coluci, VR and Legoas, SB and de Aguiar, MAM and Galvao, DS},
url = {http://iopscience.iop.org/0957-4484/16/4/041},
year = {2005},
date = {2005-01-01},
journal = {Nanotechnology},
volume = {16},
number = {4},
pages = {583},
publisher = {IOP Publishing},
abstract = {The motion of coupled oscillators based on multiwalled carbon nanotubes is studied using rigid-body dynamics simulations. The results show the existence of chaotic and regular behaviours for a given total energy, indicating the manifestation of chaos in nanoscaled mechanical systems based on carbon nanotube oscillators. Different regular motions are observed for different total energies, and they can be obtained by appropriately choosing the initial conditions. This possibility can allow the construction of multi-functional nano-devices based on multiwalled carbon nanotube oscillators.},
keywords = {Carbon Nanotubes, Chaos, Oscillators},
pubstate = {published},
tppubtype = {article}
}
Sato, F; Moreira, AS; Coura, PZ; Dantas, SO; Legoas, SB; Ugarte, D; Galvao, DS
Computer simulations of gold nanowire formation: the role of outlayer atoms Journal Article
In: Applied Physics A (invited paper), vol. 81, no. 8, pp. 1527–1531, 2005.
Abstract | Links | BibTeX | Tags: Linear Atomic Chains, Metallic Nanowires, Molecular Dynamics, TEM
@article{sato2005computer,
title = {Computer simulations of gold nanowire formation: the role of outlayer atoms},
author = {Sato, F and Moreira, AS and Coura, PZ and Dantas, SO and Legoas, SB and Ugarte, D and Galvao, DS},
url = {http://link.springer.com/article/10.1007/s00339-005-3390-2},
year = {2005},
date = {2005-01-01},
journal = {Applied Physics A (invited paper)},
volume = {81},
number = {8},
pages = {1527--1531},
publisher = {Springer-Verlag},
abstract = {Metallic nanowires (NWs) have been the object of intense theoretical and experimental investigations in the last years. In this work we present and review a new methodology we developed to study NW formation from mechanical stretching. This methodology is based on tight-binding molecular dynamics techniques using second-moment approximations. This methodology had been proven to be very effective in the study of NWs, reliably reproducing the main experimentally observed structural features. We have also investigated the problem of determining from what regions the atoms composing the linear atomic chains come. Our results show that ∼90% of these atoms come from outmost external layers.
},
keywords = {Linear Atomic Chains, Metallic Nanowires, Molecular Dynamics, TEM},
pubstate = {published},
tppubtype = {article}
}
Legoas, SB; Rodrigues, V; Ugarte, D; Galvao, DS
Legoas et al. Reply Journal Article
In: Physical Review Letters, vol. 95, no. 16, pp. 169602, 2005.
Links | BibTeX | Tags: Linear Atomic Chains, Metallic Nanowires, Molecular Dynamics
@article{legoas2005legoas,
title = {Legoas et al. Reply},
author = {Legoas, SB and Rodrigues, V and Ugarte, D and Galvao, DS},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.95.169602},
year = {2005},
date = {2005-01-01},
journal = {Physical Review Letters},
volume = {95},
number = {16},
pages = {169602},
keywords = {Linear Atomic Chains, Metallic Nanowires, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Giro, Ronaldo; Caldas, Mar'ilia Junqueira; Galvao, Douglas Soares
Band gap engineering for poly (p-phenylene) and poly (p-phenylene vinylene) copolymers using the tight-binding approach Journal Article
In: International Journal of Quantum Chemistry, vol. 103, no. 5, pp. 588–596, 2005.
Abstract | Links | BibTeX | Tags: Conducting Polymers, Electronic Structure, PPP, PPV
@article{giro2005band,
title = {Band gap engineering for poly (p-phenylene) and poly (p-phenylene vinylene) copolymers using the tight-binding approach},
author = {Giro, Ronaldo and Caldas, Mar'ilia Junqueira and Galvao, Douglas Soares},
url = {http://onlinelibrary.wiley.com/doi/10.1002/qua.20551/full},
year = {2005},
date = {2005-01-01},
journal = {International Journal of Quantum Chemistry},
volume = {103},
number = {5},
pages = {588--596},
publisher = {Wiley Online Library},
abstract = {The interest in poly(p-phenylene) (PPP) and poly(p-phenylene vinylene) (PPV) copolymers stems from the fact that these homopolymers present interesting optical and electronic properties that allow a great variety of technological applications. Combining different numbers of PPP and PPV units it is possible, in principle, to obtain new structures presenting intermediate gap values (2.8 eV and 2.4 eV for PPP and PPV, respectively). For this study we used a Hückel Hamiltonian tight-binding coupled to the negative factor counting (NFC) technique. We carried out a systematic search to determine optimum relative concentrations for disordered binary polymeric alloys with predefined gap values. Once these structures were obtained, we used the semiempirical methods AM1/PM3 and ZINDO/S-CI for geometrical and optical studies, respectively. Our theoretical results show that it is possible to obtain copolymers of PPP and PPV with intermediate gap values of their parent structures. © 2005 Wiley Periodicals, Inc. Int J Quantum Chem, 2005},
keywords = {Conducting Polymers, Electronic Structure, PPP, PPV},
pubstate = {published},
tppubtype = {article}
}
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ

