
M, Ajayan Pulickel; Woellner, Cristiano F; Owuor, Peter S; Trigueiro, Joao P C; Machado, Leonardo D; Silva, Wellington M; Kosolwattana, Suppanat; Jaques, Ygor M; Silva, Carlos J R; Pedrotti, Jairo; Tiwary, Chandra S; Chipara, Alin C; Galvao, Douglas; Chopra, Nitin; Odeh, Ihab N; Silva, Glaura G.
Hybrid 2D Nanostructures for Mechanical Reinforcement and Thermal Conductivity Enhancement in Polymer Composite Journal Article
In: Composites Science and Technology, vol. 159, no. 5, pp. 103-110, 2018.
@article{M2018,
title = {Hybrid 2D Nanostructures for Mechanical Reinforcement and Thermal Conductivity Enhancement in Polymer Composite},
author = {Ajayan Pulickel M and Cristiano F Woellner and Peter S Owuor and Joao P C Trigueiro and Leonardo D Machado and Wellington M Silva and Suppanat Kosolwattana and Ygor M Jaques and Carlos J R Silva and Jairo Pedrotti and Chandra S Tiwary and Alin C Chipara and Douglas Galvao and Nitin Chopra and Ihab N Odeh and Glaura G. Silva
},
doi = {https://doi.org/10.1016/j.compscitech.2018.01.032},
year = {2018},
date = {2018-01-01},
journal = {Composites Science and Technology},
volume = {159},
number = {5},
pages = {103-110},
abstract = {Hexagonal boron nitride (h-BN), graphene oxide (GO) and hybrid (GO/h-BN) nanosheets were employed as fillers in order to enhance the physical properties of the polymer matrix. Composites based in epoxy and these two-dimensional (2D) nanofillers were produced with different wt% and their microstructure, mechanical and thermal properties were investigated. Increases up to 140% in tensile strength, 177% in ultimate strain and 32% in elastic modulus were observed for the hybrid GO/h-BN composite with 0.5 wt% content. The hybrid nanofiller also contributed to the increase up to 142% on thermal conductivity with respect to the pure epoxy for GO/h-BN composite with 2.0 wt% content. Molecular dynamic simulation was used to predict the behavior of possible stacking arrangements between h-BN and GO nanosheets tensioned by normal and shear forces. The results showed that the hybrid GO/h-BN combination can prevent the re-stacking process of exfoliated layers, demonstrating the synergism between these nanostructures with the final effect of better dispersion in the composite material. The excellent thermal and mechanical performance of these hybrid composites en- gineered by the combination of different types of the 2D inorganic nanoparticles make them multifunctional candidates for advanced materials applications.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Oliveira, Eliezer Fernando; da Silva Autreto, Pedro Alves; Galvao, Douglas Soares
Silver Hardening via Hypersonic Impacts Journal Article
In: MRS Advances, vol. 3, no. 8-9, pp. 489-494, 2018.
@article{Oliveira2018b,
title = {Silver Hardening via Hypersonic Impacts},
author = {Eliezer Fernando Oliveira and Pedro Alves da Silva Autreto and Douglas Soares Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/silver-hardening-via-hypersonic-impacts/6A35FAB117B4FD244BBD11A64CD25160},
doi = {DOI: 10.1557/adv.2018. 173},
year = {2018},
date = {2018-01-01},
journal = {MRS Advances},
volume = {3},
number = {8-9},
pages = {489-494},
abstract = {The search for new ultra strong materials has been a very active research area. With relation to metals, a successful way to improve their strength is by the creation of a gradient of nanograins (GNG) inside the material. Recently, R. Thevamaran et al. [Science v354, 312- 316 (2016)] propose a single step method based on high velocity impact of silver nanocubes to produce high-quality GNG. This method consists of producing high impact collisions of silver cubes at hypersonic velocity (~400 m/s) against a rigid wall. Although they observed an improvement in the mechanical properties of the silver after the impact, the GNG creation and the strengthening mechanism at nanoscale remain unclear. In order to gain further insights about these mechanisms, we carried out fully atomistic molecular dynamics simulations (MD) to investigate the atomic conformations/rearrangements during and after high impact collisions of silver nanocubes at ultrasonic velocity. Our results indicate the co- existence of polycrystalline arrangements after the impact formed by core HCP domains surrounded by FCC ones, which could also contribute to explain the structural hardening.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Oliveira, Eliezer Fernando; Paupitz, Ricardo; da Silva Autreto, Pedro Alves; Moshkalev, Stanislav; Galvao, Douglas Soares
Improving Graphene-metal Contacts: Thermal Induced Polishing Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 73-78, 2018.
@article{Oliveira2018c,
title = {Improving Graphene-metal Contacts: Thermal Induced Polishing },
author = {Eliezer Fernando Oliveira and Ricardo Paupitz and Pedro Alves da Silva Autreto and Stanislav Moshkalev and Douglas Soares Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/improving-graphenemetal-contacts-thermal-induced-polishing/AC01C4996B90B0EE5E03220604071D12},
doi = {https://doi.org/10.1557/adv.2018.66},
year = {2018},
date = {2018-01-01},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = {73-78},
abstract = {Graphene is a very promising material for nanoelectronics applications due to its unique and remarkable electronic and thermal properties. However, when deposited on metallic electrodes the overall thermal conductivity is significantly decreased. This phenomenon has been attributed to the mismatch between the interfaces and contact thermal resistance. Experimentally, one way to improve the graphene/metal contact is thorough high-temperature annealing, but the detailed mechanisms behind these processes remain unclear. In order to address these questions, we carried out fully atomistic reactive molecular dynamics simulations using the ReaxFF force field to investigate the interactions between multi-layer graphene and metallic electrodes (nickel) under (thermal) annealing. Our results show that the annealing induces an upward-downward movement of the graphene layers, causing a pile-driver-like effect over the metallic surface. This graphene induced movements cause a planarization (thermal polishing-like effect) of the metallic surface, which results in the increase of the effective graphene/metal contact area. This can also explain the experimentally observed improvements of the thermal and electric conductivities.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Leonardo D Machado Cristiano F Woellner, Pedro AS Autreto; Galvao, Douglas S
Structural Transformations of Carbon and Boron Nitride Nanoscrolls at High Impact Collisions Online
2017, (preprint ArXiv:1711.00378).
@online{Woellner2017,
title = {Structural Transformations of Carbon and Boron Nitride Nanoscrolls at High Impact Collisions},
author = {Cristiano F Woellner, Leonardo D Machado, Pedro AS Autreto, Jose M de Sousa, and Douglas S Galvao},
url = {https://arxiv.org/pdf/1711.00378.pdf},
year = {2017},
date = {2017-11-01},
abstract = {The behavior of nanostructures under high strain-rate conditions has been object of theoretical and experimental investigations in recent years. For instance, it has been shown that carbon and boron nitride nanotubes can be unzipped into nanoribbons at high velocity impacts. However, the response of many nanostructures to high strain-rate conditions is still not completely understood. In this work we have investigated through fully atomistic reactive (ReaxFF) molecular dynamics (MD) simulations the mechanical behavior of carbon (CNS) and boron nitride nanoscrolls (BNS) colliding against solid targets at high velocities,. CNS (BNS) nanoscrolls are graphene (boron nitride) membranes rolled up into papyrus-like
structures. Their open-ended topology leads to unique properties not found in close-ended analogues, such as nanotubes.Our results show that the collision products are mainly determined by impact velocities and by two impact angles, which
define the position of the scroll (i) axis and (ii) open edge relative to the target. Our MD results showed that for appropriate velocities and orientations large-scale deformations and nanoscroll fracture can occur. We also observed unscrolling (scrolls going back to quasi-planar membranes), scroll unzipping into nanoribbons, and significant
reconstruction due to breaking and/or formation of new chemical bonds. For particular edge orientations and velocities, conversion from open to close-ended topology is also possible, due to the fusion of nanoscroll walls.},
note = {preprint ArXiv:1711.00378},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
structures. Their open-ended topology leads to unique properties not found in close-ended analogues, such as nanotubes.Our results show that the collision products are mainly determined by impact velocities and by two impact angles, which
define the position of the scroll (i) axis and (ii) open edge relative to the target. Our MD results showed that for appropriate velocities and orientations large-scale deformations and nanoscroll fracture can occur. We also observed unscrolling (scrolls going back to quasi-planar membranes), scroll unzipping into nanoribbons, and significant
reconstruction due to breaking and/or formation of new chemical bonds. For particular edge orientations and velocities, conversion from open to close-ended topology is also possible, due to the fusion of nanoscroll walls.
Parambath M Sudeep Sruthi Radhakrishnan, Jun Hyoung Park; Ajayan, Pulickel M
Multifunctional Hybrids Based on 2D Fluorinated Graphene Oxide and Superparamagnetic Iron Oxide Nanoparticles Journal Article
In: Particle & Particle Systems Characterization, vol. 34, no. 11, pp. 1700245, 2017.
@article{Radhakrishnan2017,
title = {Multifunctional Hybrids Based on 2D Fluorinated Graphene Oxide and Superparamagnetic Iron Oxide Nanoparticles},
author = {Sruthi Radhakrishnan, Parambath M Sudeep, Jun Hyoung Park, Cristiano F Woellner, Kierstein Maladonado, Douglas S Galvao, Benny Abraham Kaipparettu, Chandra Sekhar Tiwary, and Pulickel M Ajayan},
url = {http://onlinelibrary.wiley.com/doi/10.1002/ppsc.201700245/full},
doi = {DOI: 10.1002/ppsc.201700245},
year = {2017},
date = {2017-11-01},
journal = {Particle & Particle Systems Characterization},
volume = {34},
number = {11},
pages = {1700245},
abstract = {Carbon-based nanomaterials have garnered a lot of attention in the research of yesteryear. Here this study reports a composite based on fluorinated graphene oxide—a multifunctional subsidiary of graphene; and iron oxide nanoparticles as a contrast agent for magnetic resonance imaging (MRI). Extensive structural and functional characterization is carried out to understand composite behavior toward biotoxicity and its performance as a contrast agent. The electron withdrawing fluorine group decreases the charge transfer to iron oxide increasing the magnetic saturation of the composite thus enhancing the contrast. The interaction of paramagnetic and superparamagnetic systems yields a superior contrast agent for MRI and fluorescent imaging.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Han, Yang; Zhou, Yanguang; Qin, Guangzhao; Dong, Jinming; Galvao, Douglas S; Hu, Ming
Unprecedented mechanical response of the lattice thermal conductivity of auxetic carbon crystals Journal Article
In: Carbon, vol. 122, pp. 374-380, 2017.
@article{Han2017,
title = {Unprecedented mechanical response of the lattice thermal conductivity of auxetic carbon crystals},
author = {Han, Yang and Zhou, Yanguang and Qin, Guangzhao and Dong, Jinming and Galvao, Douglas S and Hu, Ming},
url = {http://www.sciencedirect.com/science/article/pii/S0008622317306760},
doi = {10.1016/j.carbon.2017.06.100},
year = {2017},
date = {2017-10-01},
journal = {Carbon},
volume = {122},
pages = {374-380},
abstract = {Lattice thermal conductivity (κ) of bulk materials usually increases under compression and decreases under tension, while there are still some unusual systems, exhibiting reduced κ when compressed. However, to date it has never been reported for a bulk material, whose κ is substantially enhanced under tensile strain. In this paper, we have studied thermal transport of three auxetic carbon crystals: cis-C, trans-C and hin-C for short, and their strain responses by performing first-principles calculations. It is intriguing to find that their κ are much lower than those of their allotropes, and further decrease abnormally under compression. More strikingly, κ of trans-C (cis-C) anomalously increases with tensile strain up to 7% (6%) with maximum κ of almost 7 (5) times larger than the unstrained value. The abnormal strain dependent κ are attributed to the dominant role of the enhancement of phonon lifetime under stretching, which can be further explained from the unique atomic structure of the main chain of polydiacetylene in trans-C and cis-C. The weakening of phonon anharmonicity is reflected by the enhancement of root mean-square displacement values. The reported giant augmentation of κ may inspire intensive research on auxetic carbon crystals as potential materials for emerging nanoelectronic devices.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
BORGES, Daiane DAMASCENO; NORMAND, Perine; PERMIAKOVA, Anastasia; BABARAO, Ravichandar; HEYMANS, Nicolas; GALVAO, Douglas S.; SERRE, Christian; WEIRELD, Guy DE; MAURIN, Guillaume
Gas Adsorption and Separation by the Al-based Metal-Organic Framework MIL-160 Journal Article
In: Journal of Physical Chemistry C, vol. 121, no. 48, pp. 26822–26832, 2017.
@article{BORGES2017b,
title = {Gas Adsorption and Separation by the Al-based Metal-Organic Framework MIL-160},
author = {Daiane DAMASCENO BORGES and Perine NORMAND and Anastasia PERMIAKOVA and Ravichandar BABARAO and Nicolas HEYMANS and Douglas S. GALVAO and Christian SERRE and Guy DE WEIRELD and Guillaume MAURIN},
url = {http://pubs.acs.org/doi/abs/10.1021/acs.jpcc.7b08856},
doi = {DOI: 10.1021/acs.jpcc.7b08856},
year = {2017},
date = {2017-09-14},
journal = {Journal of Physical Chemistry C},
volume = {121},
number = {48},
pages = {26822–26832},
abstract = {One of the most promising technologies, with a low energy penalty, for CO2 capture from diverse gas mixtures is based on the adsorption process using adsorbents. Many efforts are still currently deployed to search for water stable porous metal–organic frameworks (MOFs) with high CO2 affinity combined with large CO2 uptake. In this context, we have selected the water stable and easily scalable Al-based MOF MIL-160 showing an ultramicroporosity and potential interacting sites (hydroxyl and furan), both features being a priori relevant to favor the selective adsorption of CO2 over other gases including H2, N2, CH4, and CO. Density functional theory (DFT) and force-field-based grand-canonical Monte Carlo (GCMC) simulations were first coupled to predict the strength of host/guest interactions and the adsorption isotherms for all guests as single components and binary mixtures. This computational approach reveals the promises of this solid for the selective adsorption of CO2 with respect to these other investigated gases, controlled by a combination of thermodynamics and confinement effects. These predicted performances were further supported by real-coadsorption measurements performed on shaped samples which indicated that MIL-160(Al) shows promising performance for the selective CO2 capture in post- and pre-combustion conditions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Sajadi, Seyed Mohammad; Owuor, Peter Samora; Schara, Steven; Woellner, Cristiano F.; Rodrigues, Varlei; Vajtai, Robert; Lou, Jun; Galvao, Douglas S.; Tiwary, Chandra Sekhar; Ajayan, Pulickel M.
Multi-scale Geometric Design Principles Applied to 3D Printed Schwartizes Journal Article
In: Advanced Materials, vol. 2017, pp. 1704820, 2017.
@article{Sajadi2017,
title = {Multi-scale Geometric Design Principles Applied to 3D Printed Schwartizes},
author = {Seyed Mohammad Sajadi and Peter Samora Owuor and Steven Schara and Cristiano F. Woellner and Varlei Rodrigues and Robert Vajtai and Jun Lou and Douglas S. Galvao and Chandra Sekhar Tiwary and Pulickel M. Ajayan},
url = {http://onlinelibrary.wiley.com/doi/10.1002/adma.201704820/full},
doi = {10.1002/adma.201704820},
year = {2017},
date = {2017-09-14},
journal = {Advanced Materials},
volume = {2017},
pages = {1704820},
abstract = {Schwartzites are 3D porous solids with periodic minimal surfaces having negative Gaussian curvatures and can possess unusual mechanical and electronic properties. The mechanical behavior of primitive and gyroid schwartzite structures across different length scales is investigated after these geometries are 3D printed at centimeter length scales based on molec- ular models. Molecular dynamics and nite elements simulations are used
to gain further understanding on responses of these complex solids under compressive loads and kinetic impact experiments. The results show that these structures hold great promise as high load bearing and impact-resistant materials due to a unique layered deformation mechanism that emerges in these architectures during loading. Easily scalable techniques such as 3D printing can be used for exploring mechanical behavior of various predicted complex geometrical shapes to build innovative engineered materials with tunable properties.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
to gain further understanding on responses of these complex solids under compressive loads and kinetic impact experiments. The results show that these structures hold great promise as high load bearing and impact-resistant materials due to a unique layered deformation mechanism that emerges in these architectures during loading. Easily scalable techniques such as 3D printing can be used for exploring mechanical behavior of various predicted complex geometrical shapes to build innovative engineered materials with tunable properties.
Manimunda, P; Nakanishi, Y; Jaques, YM; Susarla, S; Woellner, CF; Bhowmick, S; Asif, SAS; Galvao, DS; Tiwary, CS; Ajayan, PM
Nanoscale deformation and friction characteristics of atomically thin WSe2 and heterostructure using nanoscratch and Raman spectroscopy Journal Article
In: 2D Materials, vol. 4, no. 4, pp. 045005, 2017.
@article{Manimunda2017,
title = {Nanoscale deformation and friction characteristics of atomically thin WSe2 and heterostructure using nanoscratch and Raman spectroscopy},
author = {Manimunda, P and Nakanishi, Y and Jaques, YM and Susarla, S and Woellner, CF and Bhowmick, S and Asif, SAS and Galvao, DS and Tiwary, CS and Ajayan, PM},
url = {http://iopscience.iop.org/article/10.1088/2053-1583/aa8475/meta},
doi = {10.1088/2053-1583/aa8475},
year = {2017},
date = {2017-08-23},
journal = {2D Materials},
volume = {4},
number = {4},
pages = {045005},
abstract = {2D transition metals di-selenides are attracting a lot of attention due to their interesting optical, chemical and electronics properties. Here, the deformation characteristics of monolayer, multi- layer WSe2 and its heterostructure with MoSe2 were investigated using a new technique that combines nanoscratch and Raman spectroscopy. The 2D monolayer WSe2 showed anisotropy in deformation. Effect of number of WSe2 layers on friction characteristics were explored in detail. Experimental observations were further supported by MD simulations. Raman spectra recorded from the scratched regions showed strain induced degeneracy splitting. Further nano-scale scratch tests were extended to MoSe2–WSe2 lateral heterostructures. Effect of deformation on lateral hetero junctions were further analysed using PL and Raman spectroscopy. This new technique is completely general and can be applied to study other 2D materials.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Owuor, Peter Samora; Park, Ok-Kyung; Woellner, Cristiano F; Jalilov, Almaz S; Susarla, Sandhya; Joyner, Jarin; Ozden, Sehmus; Duy, LuongXuan; Villegas Salvatierra, Rodrigo; Vajtai, Robert; Tour, James M; Lou, Jun; Galvao, Douglas S; Tiwary, Chandra S; Ajayan, P M
Lightweight Hexagonal Boron Nitride Foam for CO2 Absorption Journal Article
In: ACS Nano, vol. 11, no. 8, pp. 8944–8952, 2017.
@article{Owuor2017b,
title = {Lightweight Hexagonal Boron Nitride Foam for CO2 Absorption},
author = {Owuor, Peter Samora and Park, Ok-Kyung and Woellner, Cristiano F and Jalilov, Almaz S and Susarla, Sandhya and Joyner, Jarin and Ozden, Sehmus and Duy, LuongXuan and Villegas Salvatierra, Rodrigo and Vajtai, Robert and Tour, James M and Lou, Jun and Galvao, Douglas S and Tiwary, Chandra S and Ajayan, P M},
url = {http://pubs.acs.org/doi/abs/10.1021/acsnano.7b03291},
doi = {10.1021/acsnano.7b03291},
year = {2017},
date = {2017-08-03},
journal = {ACS Nano},
volume = {11},
number = {8},
pages = {8944–8952},
abstract = {Weak van der Waals forces between inert hexagonal boron nitride (h-BN) nanosheets make it easy for them to slide over each other, resulting in an unstable structure in macroscopic dimensions. Creating interconnections between these inert nanosheets can remarkably enhance their mechanical properties. However, controlled design of such interconnections remains a fundamental problem for many applications of h-BN foams. In this work, a scalable in situ freeze-drying synthesis of low-density, lightweight 3D macroscopic structures made of h-BN nanosheets chemically connected by poly(vinyl alcohol) (PVA) molecules via chemical cross-link is demonstrated. Unlike pristine h-BN foam which disintegrates upon handling after freeze-drying, h-BN/PVA foams exhibit stable mechanical integrity in addition to high porosity and large surface area. Fully atomistic simulations are used to understand the interactions between h-BN nanosheets and PVA molecules. In addition, the h-BN/PVA foam is investigated as a possible CO2 absorption and as laser irradiation protection material.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Borges, Daiane Damasceno; Woellner, Cristiano F; Autreto, Pedro AS; Galvao, Douglas S
2017, (preprint arXiv:1702.00250).
@online{Borges2017b,
title = {Insights on the mechanism of water-alcohol separation in multilayer graphene oxide membranes: entropic versus enthalpic factors},
author = {Borges, Daiane Damasceno and Woellner, Cristiano F and Autreto, Pedro AS and Galvao, Douglas S},
url = {https://arxiv.org/abs/1706.06213},
year = {2017},
date = {2017-06-19},
abstract = {Experimental evidences have shown that graphene oxide (GO) can be impermeable to liquids, vapors and gases, while it allows a fast permeation of water molecules. The understanding of filtration mechanisms came mostly from studies dedicated to water desalination, while very few works have been dedicated to distilling alcohols. In this work, we have investigated the molecular level mechanism underlying the alcohol/water separation inside GO membranes. A series of molecular dynamics and Grand-Canonical Monte Carlo simulations were carried out to probe the ethanol/water and methanol/water separation through GO membranes composed of multiple layered graphene-based sheets with different interlayer distance values and number of oxygen-containing functional groups. Our results show that the size exclusion and membrane affinities are not sufficient to explain the selectivity. Besides that, the favorable water molecular arrangement inside GO 2D-channels forming a robust H-bond network and the fast water diffusion are crucial for an effective separation mechanism. In other words, the separation phenomenon is not only governed by affinities with the membrane (enthalpic mechanisms) but mainly by the geometry and size factors (entropic mechanisms). We verified that the 2D geometry channel with optimal interlayer distance are key factors for designing more efficient alcohol-water separation membranes. Our findings are consistent with the available experimental data and contribute to clarify important aspects of the separation behavior of confined alcohol/water in GO membranes.},
note = {preprint arXiv:1702.00250},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Miyazaki, Celina M; Maria, Marco AE; Borges, Daiane Damasceno; Woellner, Cristiano F; Brunetto, Gustavo; Fonseca, Alexandre F; Constantino, Carlos JL; Pereira-da-Silva, Marcelo A; de Siervo, Abner; Galvao, Douglas S; Riul Jr., Antonio
2017, (preprint arXiv:1702.00250).
@online{Miyazaki2017,
title = {Synthesis, characterization and computational simulation of graphene nanoplatelets stabilized in poly (styrene sulfonate) sodium salt},
author = {Miyazaki, Celina M and Maria, Marco AE and Borges, Daiane Damasceno and Woellner, Cristiano F and Brunetto, Gustavo and Fonseca, Alexandre F and Constantino, Carlos JL and Pereira-da-Silva, Marcelo A and de Siervo, Abner and Galvao, Douglas S and Riul Jr., Antonio},
url = {https://arxiv.org/abs/1705.10673},
year = {2017},
date = {2017-05-30},
abstract = {The production of large area interfaces and the use of scalable methods to build-up designed nanostructures generating advanced functional properties are of high interest for many materials science applications. Nevertheless, large area coverage remains a major problem for pristine graphene and here we present a hybrid, composite graphene-like material soluble in water, which can be exploited in many areas, such as energy storage, electrodes fabrication, selective membranes and biosensing. Graphene oxide (GO) was produced by the traditional Hummers method being further reduced in the presence of poly(styrene sulfonate) sodium salt (PSS), thus creating stable reduced graphene oxide (rGO) nanoplateles wrapped by PSS (GPSS). Molecular dynamics simulations were carried out of further clarify the interactions between PSS molecules and rGO nanoplatelets, with calculations supported by FTIR analysis. The intermolecular forces between rGO nanoplatelets and PSS lead to the formation of a hybrid material (GPSS) stabilized by van der Waals forces, allowing the fabrication of high quality layer-by-layer (LbL) films with polyalillamine hydrochloride (PAH). Raman and electrical characterizations corroborated the successful modifications in the electronic structures from GO to GPSS after the chemical treatment, resulting in (PAH/GPSS) LbL films four orders of magnitude more conductive than (PAH/GO).
},
note = {preprint arXiv:1702.00250},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Bizao, Rafael A; Botari, Tiago; Perim, Eric; Pugno, Nicola M; Galvao, Douglas S
Mechanical properties and fracture patterns of graphene (graphitic) nanowiggles Journal Article
In: Carbon, vol. 119, pp. 431-437, 2017, (See also ArxIv version: https://arxiv.org/abs/1702.01100).
@article{Bizao2017b,
title = {Mechanical properties and fracture patterns of graphene (graphitic) nanowiggles},
author = {Bizao, Rafael A and Botari, Tiago and Perim, Eric and Pugno, Nicola M and Galvao, Douglas S},
url = {http://www.sciencedirect.com/science/article/pii/S0008622317303743},
doi = {10.1016/j.carbon.2017.04.018},
year = {2017},
date = {2017-04-14},
journal = {Carbon},
volume = {119},
pages = {431-437},
abstract = {Graphene nanowiggles (GNW) are graphene-based nanostructures obtained by making alternated regular cuts in pristine graphene nanoribbons. GNW were recently synthesized and it was demonstrated that they exhibit tunable electronic and magnetic properties by just varying their shape. Here, we have investigated the mechanical properties and fracture patterns of a large number of GNW of different shapes and sizes using fully atomistic reactive molecular dynamics simulations. Our results show that the GNW mechanical properties are strongly dependent on its shape and size and, as a general trend narrow sheets have larger ultimate strength and Young's modulus than wide ones. The estimated Young's modulus values were found to be in a range of ≈100−1000 GPa and the ultimate strength in a range of ≈20−110 GPa, depending on GNW shape. Also, super-ductile behavior under strain was observed for some structures.},
note = {See also ArxIv version: https://arxiv.org/abs/1702.01100},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
de Sousa, JM; Aguiar, AL; Girao, EC; Fonseca, Alexandre F; AG Filho, Souza; Galvao, Douglas S
Mechanical Properties and Fracture Patterns of Pentagraphene Membranes Online
2017, (preprint arXiv:1703.03789).
@online{deSousa2017,
title = {Mechanical Properties and Fracture Patterns of Pentagraphene Membranes},
author = {de Sousa, JM and Aguiar, AL and Girao, EC and Fonseca, Alexandre F and AG Filho, Souza and Galvao, Douglas S},
url = {https://arxiv.org/abs/1703.03789},
year = {2017},
date = {2017-03-10},
abstract = {Recently, a new two-dimensional carbon allotrope called pentagraphene (PG) was
proposed. PG exhibits mechanical and electronic interesting properties, including typical
band gap values of semiconducting materials. PG has a Cairo-tiling-like 2D lattice
of non coplanar pentagons and its mechanical properties have not been yet fully investigated.
In this work, we combined density functional theory (DFT) calculations and
reactive molecular dynamics (MD) simulations to investigate the mechanical properties
and fracture patterns of PG membranes under tensile strain. We show that PG
membranes can hold up to 20% of strain and that fracture occurs only after substantial
dynamical bond breaking and the formation of 7, 8 and 11 carbon rings and carbon
chains. The stress-strain behavior was observed to follow two regimes, one exhibiting linear elasticity followed by a plastic one, involving carbon atom re-hybridization with
the formation of carbon rings and chains. Our results also show that mechanically
induced structural transitions from PG to graphene is unlikely to occur, in contrast to
what was previously speculated in the literature.},
note = {preprint arXiv:1703.03789},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
proposed. PG exhibits mechanical and electronic interesting properties, including typical
band gap values of semiconducting materials. PG has a Cairo-tiling-like 2D lattice
of non coplanar pentagons and its mechanical properties have not been yet fully investigated.
In this work, we combined density functional theory (DFT) calculations and
reactive molecular dynamics (MD) simulations to investigate the mechanical properties
and fracture patterns of PG membranes under tensile strain. We show that PG
membranes can hold up to 20% of strain and that fracture occurs only after substantial
dynamical bond breaking and the formation of 7, 8 and 11 carbon rings and carbon
chains. The stress-strain behavior was observed to follow two regimes, one exhibiting linear elasticity followed by a plastic one, involving carbon atom re-hybridization with
the formation of carbon rings and chains. Our results also show that mechanically
induced structural transitions from PG to graphene is unlikely to occur, in contrast to
what was previously speculated in the literature.
Cristiano F Woellner Peter Samora Owuor, Tong Li
High Toughness in Ultralow Density Graphene Oxide Foam Journal Article
In: Advanced Materials Interfaces, vol. 4, no. 10, pp. 1700030, 2017.
@article{Owuor2017,
title = {High Toughness in Ultralow Density Graphene Oxide Foam},
author = {Peter Samora Owuor, Cristiano F Woellner, Tong Li, Soumya Vinod, Sehmus Ozden, Suppanat Kosolwattana, Sanjit Bhowmick, Luong Xuan Duy, Rodrigo V Salvatierra, Bingqing Wei, Syed AS Asif, James M Tour, Robert Vajtai, Jun Lou, Douglas S Galvão, Chandra Sekhar Tiwary, Pulickel Ajayan},
url = {http://onlinelibrary.wiley.com/doi/10.1002/admi.201700030/abstract },
doi = {10.1002/admi.201700030},
year = {2017},
date = {2017-03-01},
journal = {Advanced Materials Interfaces},
volume = {4},
number = {10},
pages = {1700030},
abstract = {Here, the scalable synthesis of low-density 3D macroscopic structure of graphene oxide (GO) interconnected with polydimethylsiloxane (PDMS) is reported. A controlled amount of PDMS is infused into the freeze-dried foam to result into a very rigid structure with improved mechanical properties, such as tensile plasticity and toughness. The PDMS wets the graphene oxide sheets and acts like glue between the 2D sheets. Molecular dynamics simulations are used to further elucidate the mechanisms of the interactions of graphene oxide layers with PDMS. The ability of using the interconnecting graphene oxide foam as an effective oil–water separator and stable insulating behavior to elevated temperatures are further demonstrated. The structural rigidity of the sample is also tested using laser impact and compared with GO foam.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Splugues, Vinicius; da Silva Autreto, Pedro Alves; Galvao, Douglas S
Hydrogenation Dynamics of Biphenylene Carbon (Graphenylene) Membranes Journal Article
In: MRS Advances, vol. 2017, pp. 1-6, 2017.
@article{Splugues2017,
title = {Hydrogenation Dynamics of Biphenylene Carbon (Graphenylene) Membranes},
author = {Splugues, Vinicius and da Silva Autreto, Pedro Alves and Galvao, Douglas S},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/hydrogenation-dynamics-of-biphenylene-carbon-graphenylene-membranes/139DB900D41560D64F352A31CE219D3A},
doi = {10.1557/adv.2017.239},
year = {2017},
date = {2017-02-28},
journal = {MRS Advances},
volume = {2017},
pages = {1-6},
abstract = {The advent of graphene created a revolution in materials science. Because of this there is a renewed interest in other carbon-based structures. Graphene is the ultimate (just one atom thick) membrane. It has been proposed that graphene can work as impermeable membrane to standard gases, such argon and helium. Graphene-like porous membranes, but presenting larger porosity and potential selectivity would have many technological applications. Biphenylene carbon (BPC), sometimes called graphenylene, is one of these structures. BPC is a porous two-dimensional (planar) allotrope carbon, with its pores resembling typical sieve cavities and/or some kind of zeolites. In this work, we have investigated the hydrogenation dynamics of BPC membranes under different conditions (hydrogenation plasma density, temperature, etc.). We have carried out an extensive study through fully atomistic molecular dynamics (MD) simulations using the reactive force field ReaxFF, as implemented in the well-known Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) code. Our results show that the BPC hydrogenation processes exhibit very complex patterns and the formation of correlated domains (hydrogenated islands) observed in the case of graphene hydrogenation was also observed here. MD results also show that under hydrogenation BPC structure undergoes a change in its topology, the pores undergoing structural transformations and extensive hydrogenation can produce significant structural damages, with the formation of large defective areas and large structural holes, leading to structural collapse.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Borges, Daiane Damasceno; Maurin, Guillaume; Galvao, Douglas S
Design of Porous Metal-Organic Frameworks for Adsorption Driven Thermal Batteries Journal Article
In: MRS Advances, vol. 2017, pp. 1-6, 2017.
@article{Borges2017b,
title = {Design of Porous Metal-Organic Frameworks for Adsorption Driven Thermal Batteries},
author = {Borges, Daiane Damasceno and Maurin, Guillaume and Galvao, Douglas S},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/design-of-porous-metalorganic-frameworks-for-adsorption-driven-thermal-batteries/A63B92E4D7E413D7CC047E152C7F22AF},
doi = {10.1557/adv.2017.181},
year = {2017},
date = {2017-02-15},
journal = {MRS Advances},
volume = {2017},
pages = {1-6},
abstract = {Thermal batteries based on a reversible adsorption/desorption of a working fluid (water, methanol, ammonia) rather than the conventional vapor compression is a promising alternative to exploit waste thermal energy for heat reallocation. In this context, there is an increasing interest to find novel porous solids able to adsorb a high energy density of working fluid under low relative vapor pressure condition combined with an easy ability of regeneration (desorption) at low temperature, which are the major requirements for adsorption driven heat pumps and chillers. The porous crystalline hybrid materials named Metal–Organic Frameworks (MOF) represent a great source of inspiration for sorption based-applications owing to their tunable chemical and topological features associated with a large variability of pore sizes. Recently, we have designed a new MOF named MIL-160 (MIL stands for Materials of Institut Lavoisier), isostructural to CAU-10, built from the assembly of corner sharing aluminum chains octahedra AlO4(OH)2 with the 2,5-furandicarboxylic linker substituting the pristine organic linker, 1,4-benzenedicarboxylate. This ligand replacement strategy proved to enhance both the hydrophilicity of the MOF and its amount of water adsorbed at low p/p0. This designed solid was synthesized and its chemical stability/adsorption performances verified. Here, we have extended this study by incorporating other polar heterocyclic linkers and a comparative computational study of the water adsorption performances of these novel structures has been performed. To that purpose, the cell and geometry optimizations of all hypothetical frameworks were first performed at the density functional theory level and their water adsorption isotherms were further predicted by using force-field based Grand-Canonical Monte Carlo simulations. This study reveals the ease tunable water affinity of MOF for the desired application.
},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Bizao, Rafael A; Botari, Tiago; Perim, Eric; Pugno, Nicola M; Galvao, Douglas S
Mechanical Properties and Fracture Patterns of Graphene (Graphitic) Nanowiggles Online
2017, (preprint arXiv:1702.01100).
@online{Bizao2017,
title = {Mechanical Properties and Fracture Patterns of Graphene (Graphitic) Nanowiggles},
author = {Bizao, Rafael A and Botari, Tiago and Perim, Eric and Pugno, Nicola M and Galvao, Douglas S},
url = {https://arxiv.org/pdf/1702.01100.pdf},
year = {2017},
date = {2017-02-03},
abstract = {Graphene nanowiggles (GNW) are graphene-based nanostructures
obtained by making alternated regular cuts in pristine graphene nanoribbons.
GNW were recently synthesized and it was demonstrated that
they exhibit tunable electronic and magnetic properties by just varying
their shape. Here, we have investigated the mechanical properties and
fracture patterns of a large number of GNW of different shapes and
sizes using fully atomistic reactive molecular dynamics simulations.
Our results show that the GNW mechanical properties are strongly
dependent on its shape and size and, as a general trend narrow sheets
have larger ultimate strength and Young’s modulus than wide ones.
The estimated Young’s modulus values were found to be in a range of
≈ 100 − 1000 GPa and the ultimate strength in a range of ≈ 20 − 110
GPa, depending on GNW shape. Also, super-ductile behaviour under
strain was observed for some structures.},
note = {preprint arXiv:1702.01100},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
obtained by making alternated regular cuts in pristine graphene nanoribbons.
GNW were recently synthesized and it was demonstrated that
they exhibit tunable electronic and magnetic properties by just varying
their shape. Here, we have investigated the mechanical properties and
fracture patterns of a large number of GNW of different shapes and
sizes using fully atomistic reactive molecular dynamics simulations.
Our results show that the GNW mechanical properties are strongly
dependent on its shape and size and, as a general trend narrow sheets
have larger ultimate strength and Young’s modulus than wide ones.
The estimated Young’s modulus values were found to be in a range of
≈ 100 − 1000 GPa and the ultimate strength in a range of ≈ 20 − 110
GPa, depending on GNW shape. Also, super-ductile behaviour under
strain was observed for some structures.
Borges, Daiane D; Woellner, Cristiano F; Autreto, Pedro AS; Galvao, Douglas S
2017, (preprint arXiv:1702.00250).
@online{Borges2017,
title = {Water Permeation through Layered Graphene-based Membranes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Borges, Daiane D and Woellner, Cristiano F and Autreto, Pedro AS and Galvao, Douglas S},
url = {https://arxiv.org/abs/1702.00250},
year = {2017},
date = {2017-02-01},
abstract = {Graphene-based membranes have been investigated as promising candidates for water
filtration and gas separation applications. Experimental evidences have shown that graphene
oxide can be impermeable to liquids, vapors and gases, while allowing a fast permeation of water
molecules. This phenomenon has been attributed to the formation of a network of nano
capillaries that allow nearly frictionless water flow while blocking other molecules by steric
hindrance effects. It is supposed that water molecules are transported through the percolated twodimensional
channels formed between graphene-based sheets. Although these channels allow
fast water permeation in such materials, the flow rates are strongly dependent on how the
membranes are fabricated. Also, some fundamental issues regarding the nanoscale mechanisms
of water permeation are still not fully understood and their interpretation remains controversial.
In this work, we have investigated the dynamics of water permeation through pristine graphene
and graphene oxide model membranes. We have carried out fully atomistic classical molecular
dynamics simulations of systems composed of multiple layered graphene-based sheets into
contact with a water reservoir under controlled thermodynamics conditions (e. g., by varying
temperature and pressure values). We have systematically analyzed how the transport dynamics
of the confined nanofluids depend on the interlayer distances and the role of the oxide functional
groups. Our results show the water flux is much more effective for graphene than for graphene
oxide membranes. These results are attributed to the H-bonds formation between oxide
functional groups and water, which traps the water molecules and precludes ultrafast water
transport through the nanochannels.},
note = {preprint arXiv:1702.00250},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
filtration and gas separation applications. Experimental evidences have shown that graphene
oxide can be impermeable to liquids, vapors and gases, while allowing a fast permeation of water
molecules. This phenomenon has been attributed to the formation of a network of nano
capillaries that allow nearly frictionless water flow while blocking other molecules by steric
hindrance effects. It is supposed that water molecules are transported through the percolated twodimensional
channels formed between graphene-based sheets. Although these channels allow
fast water permeation in such materials, the flow rates are strongly dependent on how the
membranes are fabricated. Also, some fundamental issues regarding the nanoscale mechanisms
of water permeation are still not fully understood and their interpretation remains controversial.
In this work, we have investigated the dynamics of water permeation through pristine graphene
and graphene oxide model membranes. We have carried out fully atomistic classical molecular
dynamics simulations of systems composed of multiple layered graphene-based sheets into
contact with a water reservoir under controlled thermodynamics conditions (e. g., by varying
temperature and pressure values). We have systematically analyzed how the transport dynamics
of the confined nanofluids depend on the interlayer distances and the role of the oxide functional
groups. Our results show the water flux is much more effective for graphene than for graphene
oxide membranes. These results are attributed to the H-bonds formation between oxide
functional groups and water, which traps the water molecules and precludes ultrafast water
transport through the nanochannels.
Solis, Daniel; Woellner, Cristiano F; Borges, Daiane D; Galvao, Douglas S
Mechanical and Thermal Stability of Graphyne and Graphdiyne Nanoscrolls Journal Article
In: MRS Advances, vol. 2017, pp. 129-134, 2017.
@article{Solis2017,
title = {Mechanical and Thermal Stability of Graphyne and Graphdiyne Nanoscrolls},
author = {Solis, Daniel and Woellner, Cristiano F and Borges, Daiane D and Galvao, Douglas S},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/mechanical-and-thermal-stability-of-graphyne-and-graphdiyne-nanoscrolls/202E7B7C471411200DE9D05C264726B8},
doi = {10.1557/adv.2017.130},
year = {2017},
date = {2017-02-01},
journal = {MRS Advances},
volume = {2017},
pages = {129-134},
abstract = {Graphynes and graphdiynes are carbon 2D allotrope structures presenting both sp2 and sp hybridized atoms. These materials have been theoretically predicted but due to intrinsic difficulties in their synthesis, only recently some of these structures have been experimentally realized. Graphyne nanoscrolls are structures obtained by rolling up graphyne sheets into papyrus-like structures. In this work, we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics of nanoscroll formation for a series of graphyne (α, β, and δ types) structures. We have also investigated their thermal stability for a temperature range of 200-1000K. Our results show that stable nanoscrolls can be formed for all structures considered here. Their stability depends on a critical value of the ratio between length and height of the graphyne sheets. Our findings also show that these structures are structurally less stable then graphene-based nanoscrolls. This can be explained by the graphyne higher structural porosity which results in a decreased pi-pi stacking interactions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2013
Autreto, PA; de Sousa, JM; Galvao, DS
On the Dynamics of Graphdiyne Hydrogenation Proceedings
Cambridge University Press, vol. 1549, 2013.
Abstract | Links | BibTeX | Tags: Graphdyine, Graphynes, Hydrogenation, Molecular Dynamics
@proceedings{autreto2013dynamics,
title = {On the Dynamics of Graphdiyne Hydrogenation},
author = {Autreto, PA and de Sousa, JM and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8915680&fileId=S1946427413006088},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1549},
pages = {59--64},
publisher = {Cambridge University Press},
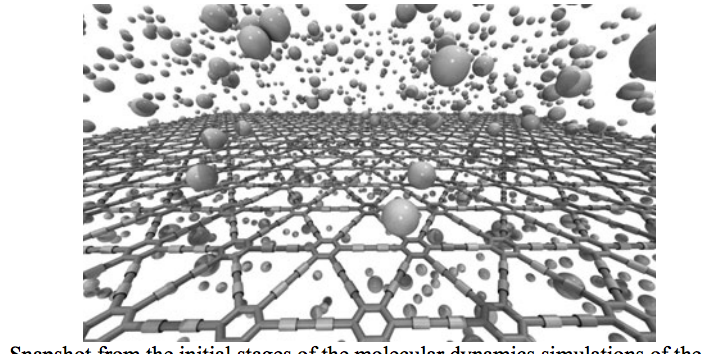
abstract = {Graphene is a two-dimensional (2D) hexagonal array of carbon atoms in sp2-hybridized states. Graphene presents unique and exceptional electronic, thermal and mechanical properties. However, in its pristine state graphene is a gapless semiconductor, which poses some limitations to its use in some transistor electronics. Because of this there is a renewed interest in other possible two-dimensional carbon-based structures similar to graphene. Examples of this are graphynes and graphdiynes, which are two-dimensional structures, composed of carbon atoms in sp2 and sp-hybridized states. Graphdiynes (benzenoid rings connecting two acetylenic groups) were recently synthesized and they can be intrinsically nonzero gap systems. These systems can be easily hydrogenated and the amount of hydrogenation can be used to tune the band gap value. In this work we have investigated, through fully atomistic molecular dynamics simulations with reactive force field (ReaxFF), the structural and dynamics aspects of the hydrogenation mechanisms of graphdiyne membranes. Our results showed that depending on whether the atoms are in the benzenoid rings or as part of the acetylenic groups, the rates of hydrogenation are quite distinct and change in time in a very complex pattern. Initially, the most probable sites to be hydrogenated are the carbon atoms forming the triple bonds, as expected. But as the amount of hydrogenation increases in time this changes and then the carbon atoms forming single bonds become the preferential sites. The formation of correlated domains observed in hydrogenated graphene is no longer observed in the case of graphdiynes. We have also carried out ab initio DFT calculations for model structures in order to test the reliability of ReaxFF calculations.},
keywords = {Graphdyine, Graphynes, Hydrogenation, Molecular Dynamics},
pubstate = {published},
tppubtype = {proceedings}
}
Perim, Eric; Paupitz, Ricardo; Autreto, PAS; Galvao, DS
The Hydrogenation Dynamics of h-BN Sheets Proceedings
Cambridge University Press, vol. 1549, 2013.
Abstract | Links | BibTeX | Tags: Boron Nitride, Hydrogenation, Molecular Dynamics, Nanotubes
@proceedings{perim2013hydrogenation,
title = {The Hydrogenation Dynamics of h-BN Sheets},
author = {Perim, Eric and Paupitz, Ricardo and Autreto, PAS and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8943477&fileId=S1946427413007938},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1549},
pages = {91--98},
publisher = {Cambridge University Press},
abstract = {Hexagonal boron nitride (h-BN), also known as white graphite, is the inorganic analogue of graphite. Single layers of both structures have been already experimentally realized.
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics of hydrogenation of h-BN single-layers membranes.
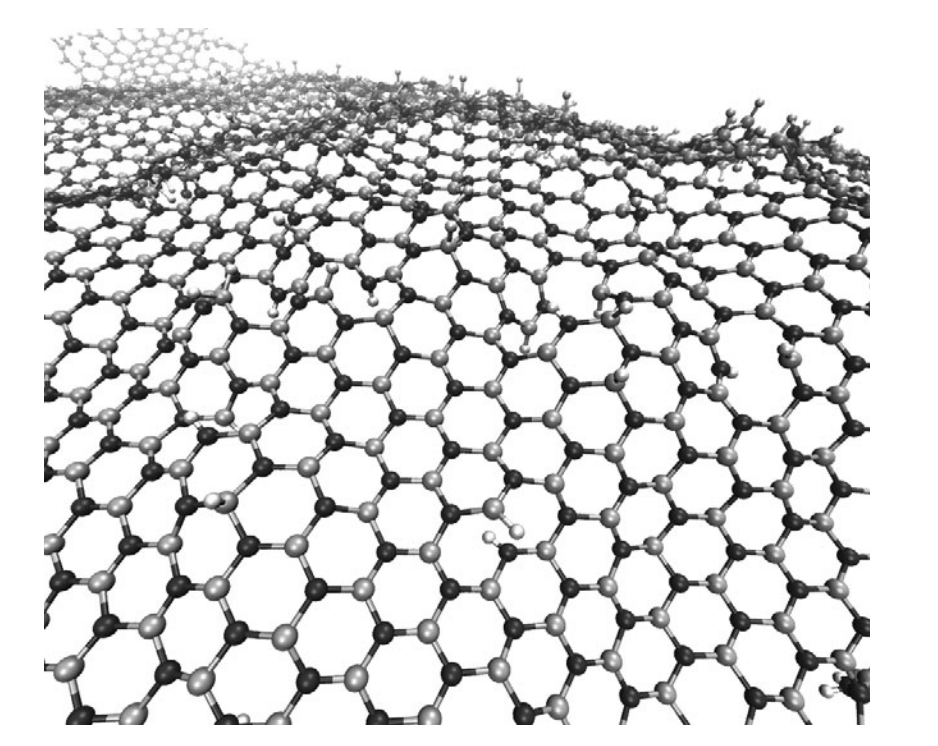
Our results show that the rate of hydrogenation atoms bonded to the membrane is highly dependent on the temperature and that only at low temperatures there is a preferential bond to boron atoms. Unlike graphanes (hydrogenated graphene), hydrogenated h-BN membranes do not exhibit the formation of correlated domains. Also, the out-of-plane deformations are more pronounced in comparison with the graphene case. After a critical number of incorporated hydrogen atoms the membrane become increasingly defective, lost its two-dimensional character and collapses. The hydrogen radial pair distribution and second-nearest neighbor correlations were also analyzed.},
keywords = {Boron Nitride, Hydrogenation, Molecular Dynamics, Nanotubes},
pubstate = {published},
tppubtype = {proceedings}
}
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics of hydrogenation of h-BN single-layers membranes.
Our results show that the rate of hydrogenation atoms bonded to the membrane is highly dependent on the temperature and that only at low temperatures there is a preferential bond to boron atoms. Unlike graphanes (hydrogenated graphene), hydrogenated h-BN membranes do not exhibit the formation of correlated domains. Also, the out-of-plane deformations are more pronounced in comparison with the graphene case. After a critical number of incorporated hydrogen atoms the membrane become increasingly defective, lost its two-dimensional character and collapses. The hydrogen radial pair distribution and second-nearest neighbor correlations were also analyzed.
Miyazaki, Celina M; Riul, Antonio; Dos Santos, David S; Ferreira, Mariselma; Constantino, Carlos JL; Pereira-da-Silva, Marcelo A; Paupitz, Ricardo; Galvao, Douglas S; others,
Bending of Layer-by-Layer Films Driven by an External Magnetic Field Journal Article
In: International journal of molecular sciences, vol. 14, no. 7, pp. 12953–12969, 2013.
Abstract | Links | BibTeX | Tags: LB films, Nanoscale Effects
@article{miyazaki2013bending,
title = {Bending of Layer-by-Layer Films Driven by an External Magnetic Field},
author = {Miyazaki, Celina M and Riul, Antonio and Dos Santos, David S and Ferreira, Mariselma and Constantino, Carlos JL and Pereira-da-Silva, Marcelo A and Paupitz, Ricardo and Galvao, Douglas S and others},
url = {http://www.mdpi.com/1422-0067/14/7/12953/htm},
year = {2013},
date = {2013-01-01},
journal = {International journal of molecular sciences},
volume = {14},
number = {7},
pages = {12953--12969},
publisher = {Multidisciplinary Digital Publishing Institute},
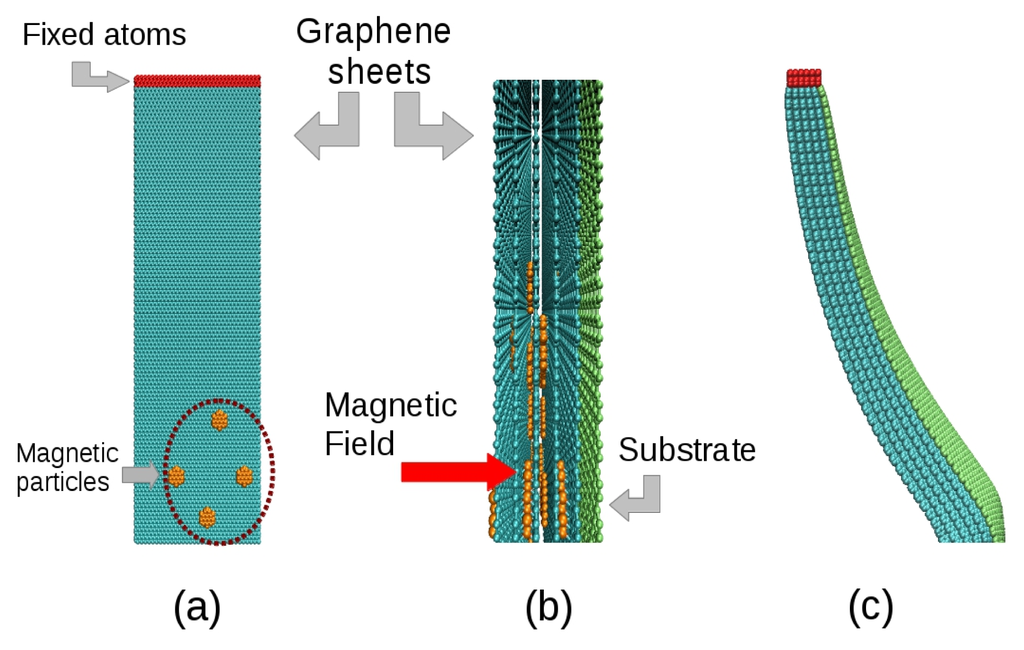
abstract = {We report on optimized architectures containing layer-by-layer (LbL) films of natural rubber latex (NRL), carboxymethyl-chitosan (CMC) and magnetite (Fe3O4) nanoparticles (MNPs) deposited on flexible substrates, which could be easily bent by an external magnetic field. The mechanical response depended on the number of deposited layers and was explained semi-quantitatively with a fully atomistic model, where the LbL film was represented as superposing layers of hexagonal graphene-like atomic arrangements deposited on a stiffer substrate. The bending with no direct current or voltage being applied to a supramolecular structure containing biocompatible and antimicrobial materials represents a proof-of-principle experiment that is promising for tissue engineering applications in biomedicine. - See more at: http://www.mdpi.com/1422-0067/14/7/12953/htm#sthash.cSUOvaot.dpuf},
keywords = {LB films, Nanoscale Effects},
pubstate = {published},
tppubtype = {article}
}
Machado, LD; Autreto, PAS; Galvao, DS
Graphyne Oxidation: Insights From a Reactive Molecular Dynamics Investigation Proceedings
Cambridge University Press, vol. 1549, 2013.
Abstract | Links | BibTeX | Tags: Graphdyine, Graphyne, Molecular Dynamics, Oxidation
@proceedings{machado2013graphyne,
title = {Graphyne Oxidation: Insights From a Reactive Molecular Dynamics Investigation},
author = {Machado, LD and Autreto, PAS and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8963025&fileId=S194642741300941X},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1549},
pages = {53--58},
publisher = {Cambridge University Press},
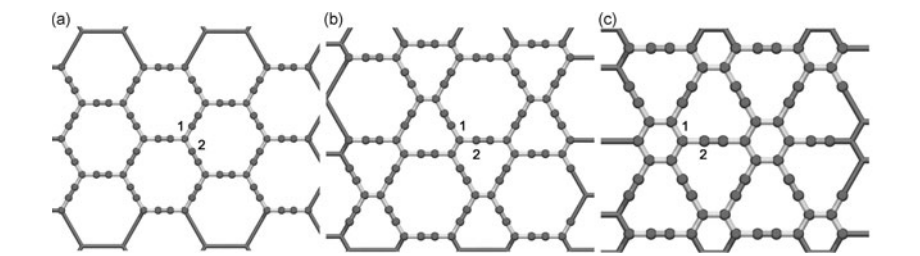
abstract = {Graphyne is a generic name for a family of carbon allotrope two-dimensional structures where sp2 (single and double bonds) and sp (triple bonds) hybridized states coexists. They exhibit very interesting electronic and mechanical properties sharing some of the unique graphene characteristics. Similarly to graphene, the graphyne electronic properties can be modified by chemical functionalization, such as; hydrogenation, fluorination and oxidation. Oxidation is of particular interest since it can produce significant structural damages.
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics and structural changes of the oxidation of single-layer graphyne membranes at room temperature. We have considered α, β, and γ-graphyne structures. Our results showed that the oxidation reactions are strongly site dependent and that the sp-hybridized carbon atoms are the preferential sites to chemical attacks. Our results also showed that the effectiveness of the oxidation (estimated from the number of oxygen atoms covalently bonded to carbon atoms) follows the α, β, γ-graphyne structure ordering. These differences can be explained by the fact that for α-graphyne structures the oxidation reactions occur in two steps: first, the oxygen atoms are trapped at the center of the large polygonal rings and then they react with the carbon atoms composing of the triple bonds. The small rings of γ-graphyne structures prevent these reactions to occur. The effectiveness of β-graphyne oxidation is between the α- and γ-graphynes.},
keywords = {Graphdyine, Graphyne, Molecular Dynamics, Oxidation},
pubstate = {published},
tppubtype = {proceedings}
}
In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics and structural changes of the oxidation of single-layer graphyne membranes at room temperature. We have considered α, β, and γ-graphyne structures. Our results showed that the oxidation reactions are strongly site dependent and that the sp-hybridized carbon atoms are the preferential sites to chemical attacks. Our results also showed that the effectiveness of the oxidation (estimated from the number of oxygen atoms covalently bonded to carbon atoms) follows the α, β, γ-graphyne structure ordering. These differences can be explained by the fact that for α-graphyne structures the oxidation reactions occur in two steps: first, the oxygen atoms are trapped at the center of the large polygonal rings and then they react with the carbon atoms composing of the triple bonds. The small rings of γ-graphyne structures prevent these reactions to occur. The effectiveness of β-graphyne oxidation is between the α- and γ-graphynes.

Perim, E; Autreto, PAS; Paupitz, R; Galvao, DS
Dynamical aspects of the unzipping of multiwalled boron nitride nanotubes Journal Article
In: Physical Chemistry Chemical Physics, vol. 15, no. 44, pp. 19147–19150, 2013.
Abstract | Links | BibTeX | Tags: Boron Nitride, Mechanical Properties, Molecular Dynamics, Unzipping
@article{perim2013dynamical,
title = {Dynamical aspects of the unzipping of multiwalled boron nitride nanotubes},
author = {Perim, E and Autreto, PAS and Paupitz, R and Galvao, DS},
url = {http://pubs.rsc.org/EN/content/articlehtml/2013/cp/c3cp52701h},
year = {2013},
date = {2013-01-01},
journal = {Physical Chemistry Chemical Physics},
volume = {15},
number = {44},
pages = {19147--19150},
publisher = {Royal Society of Chemistry},
abstract = {Boron nitride nanoribbons (BNNRs) exhibit very interesting magnetic properties, which could be very useful in the development of spintronic based devices. One possible route to obtain BNNRs is through the unzipping of boron nitride nanotubes (BNNTs), which have been already experimentally realized. In this work, different aspects of the unzipping process of BNNTs were investigated through fully atomistic molecular dynamics simulations using a classical reactive force field (ReaxFF). We investigated multiwalled BNNTs of different diameters and chiralities. Our results show that chirality plays a very important role in the unzipping process, as well as the interlayer coupling. These combined aspects significantly change the fracturing patterns and several other features of the unzipping processes in comparison to the ones observed for carbon nanotubes. Also, similar to carbon nanotubes, defective BNNTs can create regions of very high curvature which can act as a path to the unzipping process.
},
keywords = {Boron Nitride, Mechanical Properties, Molecular Dynamics, Unzipping},
pubstate = {published},
tppubtype = {article}
}
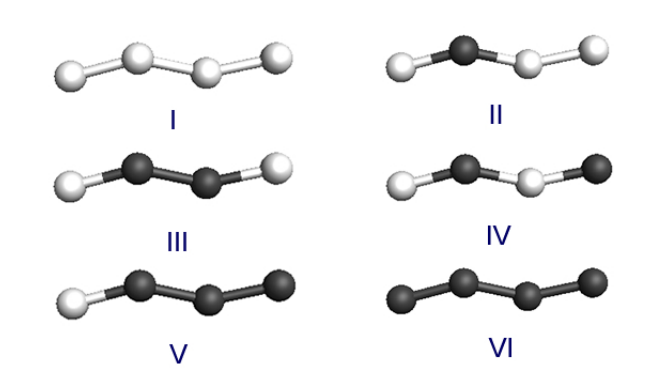
Autreto, Pedro Alves da Silva; Galvao, Douglas S; Artacho, Emilio
Species Fractionation in Atomic Chains from Mechanically Stretched Alloys Journal Article
In: arXiv preprint arXiv:1312.1285, 2013.
Abstract | Links | BibTeX | Tags: Atomic Chains, DFT, Mech, Mechanical Properties, Metallic Nanowires
@article{autreto2013species,
title = {Species Fractionation in Atomic Chains from Mechanically Stretched Alloys},
author = {Autreto, Pedro Alves da Silva and Galvao, Douglas S and Artacho, Emilio},
url = {http://arxiv.org/abs/1312.1285},
year = {2013},
date = {2013-01-01},
journal = {arXiv preprint arXiv:1312.1285},
abstract = {Bettini et al. [Nature Nanotech 1, 182 (2006)] reported the first experimental realization of linear
atomic chains (LACs) composed of different atoms (Au and Ag). Different contents of Au and Ag
were observed in the chains from what found in the bulk alloys, which rises the question of what is the
wire composition if in equilibrium with a bulk alloy. In this work we address the thermodynamic
driving force for species fractionation in LACs under tension, and we present density-functional
theory results for Ag-Au chain alloys. A pronounced stabilization of wires with an alternating
Ag-Au sequence is observed, which could be behind the experimentally observed Au enrichment in
LACs from alloys of high Ag content.},
keywords = {Atomic Chains, DFT, Mech, Mechanical Properties, Metallic Nanowires},
pubstate = {published},
tppubtype = {article}
}
atomic chains (LACs) composed of different atoms (Au and Ag). Different contents of Au and Ag
were observed in the chains from what found in the bulk alloys, which rises the question of what is the
wire composition if in equilibrium with a bulk alloy. In this work we address the thermodynamic
driving force for species fractionation in LACs under tension, and we present density-functional
theory results for Ag-Au chain alloys. A pronounced stabilization of wires with an alternating
Ag-Au sequence is observed, which could be behind the experimentally observed Au enrichment in
LACs from alloys of high Ag content.
2012
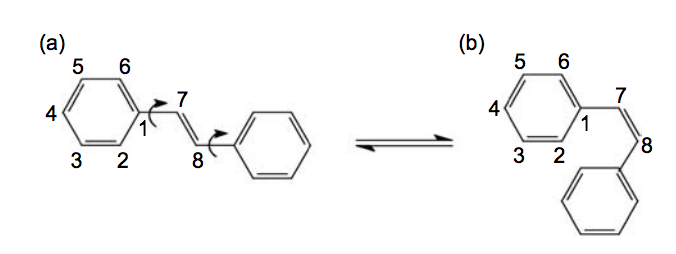
Camilo Jr, A; dos Santos, RPB; Coluci, VR; Galvao, DS
Comparative parametric method 6 (PM6) and Recife model 1 (RM1) study of trans-stilbene Journal Article
In: Molecular Simulation, vol. 38, no. 1, pp. 1–7, 2012.
Abstract | Links | BibTeX | Tags: AM1, MOPAC, PM3, PM6, PPV, RM1, Stilbene
@article{camilo2012comparative,
title = {Comparative parametric method 6 (PM6) and Recife model 1 (RM1) study of trans-stilbene},
author = {Camilo Jr, A and dos Santos, RPB and Coluci, VR and Galvao, DS},
url = {http://www.tandfonline.com/doi/abs/10.1080/08927022.2011.597392#.VLZyQ4rF-2o},
year = {2012},
date = {2012-01-01},
journal = {Molecular Simulation},
volume = {38},
number = {1},
pages = {1--7},
publisher = {Taylor & Francis Group},
abstract = {In this paper, we report a comparative parametric method 6 (PM6) and Recife model 1 (RM1) study of trans-stilbene in its ground and (excited) singlet, triplet and ionic (positive and negative polarons and bipolarons) states. We evaluated the accuracy of the recently developed PM6 and RM1 comparing the obtained results with other semi-empirical, ab initio methods and available experimental data. PM6 and RM1 predict non-planar ground and singlet states for trans-stilbene, in agreement with the PM5 and the Austin model 1. On the other hand, the PM3 predicts planar configurations, which is in agreement with the available experimental data. PM6 and RM1 overestimate the cis–trans isomerisation energy as well as the ionisation potential of both cis- and trans-stilbene. In spite of the developments of these new methods, PM3 continues to be the only one of these methods to correctly predict the conformation of stilbene.},
keywords = {AM1, MOPAC, PM3, PM6, PPV, RM1, Stilbene},
pubstate = {published},
tppubtype = {article}
}
Autreto, PAS; Galvao, Douglas S; Santos, Ricardo PB; Legoas, SB
Graphene to Fluorographene: A Reactive Molecular Dynamics Study Journal Article
In: Physicæ Proceedings, vol. 1, no. 1, pp. 3, 2012.
Abstract | Links | BibTeX | Tags: Graphanes, Graphene, Molecular Dynamics
@article{autreto2012graphene,
title = {Graphene to Fluorographene: A Reactive Molecular Dynamics Study},
author = {Autreto, PAS and Galvao, Douglas S and Santos, Ricardo PB and Legoas, SB},
url = {http://physicae.ifi.unicamp.br/phyproceedings/article/view/physicae.proceedings.XIYRM.11},
year = {2012},
date = {2012-01-01},
journal = {Physicæ Proceedings},
volume = {1},
number = {1},
pages = {3},
abstract = {We have investigated, using fully reactive molecular dynamics methodology, the structural and dynamical aspects of the fluorination of graphene membranes leading to fluographene formation. The strong and fast chemical reactivity processes involving fluorine produce distinct aspects of the observed in the case of the hydrogenation of graphene (the so called graphane formation). Fluorination tends to produce significant defective areas on the graphene membrane with alteration on the typical carbon-carbon distances, sometimes with the presence of large holes due to carbon losses. This may explain the broad distribution of values of lattice parameter experimentally observed.
},
keywords = {Graphanes, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Perim, E; Galvao, DS
Boron Nitride Nanoscrolls Journal Article
In: Physicæ Proceedings, vol. 1, no. 1, pp. 2, 2012.
Abstract | Links | BibTeX | Tags: Boron Nitride, Molecular Dynamics, Scrolls
@article{perim2012boron,
title = {Boron Nitride Nanoscrolls},
author = {Perim, E and Galvao, DS},
url = {http://physicae.ifi.unicamp.br/phyproceedings/article/view/269},
year = {2012},
date = {2012-01-01},
journal = {Physicæ Proceedings},
volume = {1},
number = {1},
pages = {2},
abstract = {Recently, based on computer simulations, it has been proposed that stable boron nitride nanoscrolls (BNNSs) can exist. In this work we show that the BNNSs stability mechanisms follow the same simple physical principles proposed for carbon nanoscrolls (CNSs). For both classes of scrolls, the mechanical stability arises as the result of the interplay between attractive van der Waals forces and the elastic (bending) deformations. The topology (chirality) of the scrolled single-layer membranes plays an important role defining BNNS stability. A controled way to produce BNNSs is also addressed.},
keywords = {Boron Nitride, Molecular Dynamics, Scrolls},
pubstate = {published},
tppubtype = {article}
}
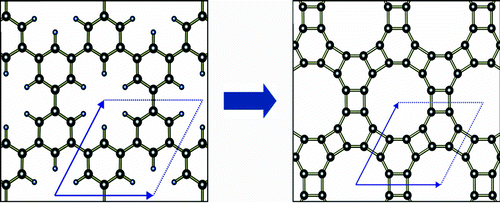
Brunetto, Gustavo; Autreto, PAS; Machado, Leonardo Dantas; Santos, BI; dos Santos, Ricardo PB; Galvao, Douglas S
Nonzero gap two-dimensional carbon allotrope from porous graphene Journal Article
In: The Journal of Physical Chemistry C, vol. 116, no. 23, pp. 12810–12813, 2012.
Abstract | Links | BibTeX | Tags: BPC, DFT, Graphene, Porous Graphene
@article{brunetto2012nonzero,
title = {Nonzero gap two-dimensional carbon allotrope from porous graphene},
author = {Brunetto, Gustavo and Autreto, PAS and Machado, Leonardo Dantas and Santos, BI and dos Santos, Ricardo PB and Galvao, Douglas S},
url = {http://pubs.acs.org/doi/abs/10.1021/jp211300n},
year = {2012},
date = {2012-01-01},
journal = {The Journal of Physical Chemistry C},
volume = {116},
number = {23},
pages = {12810--12813},
publisher = {American Chemical Society},
abstract = {Graphene is considered one of the most promising materials for future electronics. However, in its pristine form, graphene is a gapless material, which imposes limitations to its use in some electronic applications. To solve this problem, many approaches have been tried, such as physical and chemical functionalizations. These processes compromise some of the desirable graphene properties. In this work, based on ab initio quantum molecular dynamics, we showed that a two-dimensional carbon allotrope, named biphenylene carbon (BPC), can be obtained from selective dehydrogenation of porous graphene. BPC presents a nonzero bandgap and well-delocalized frontier orbitals. Synthetic routes to BPC are also addressed.},
keywords = {BPC, DFT, Graphene, Porous Graphene},
pubstate = {published},
tppubtype = {article}
}
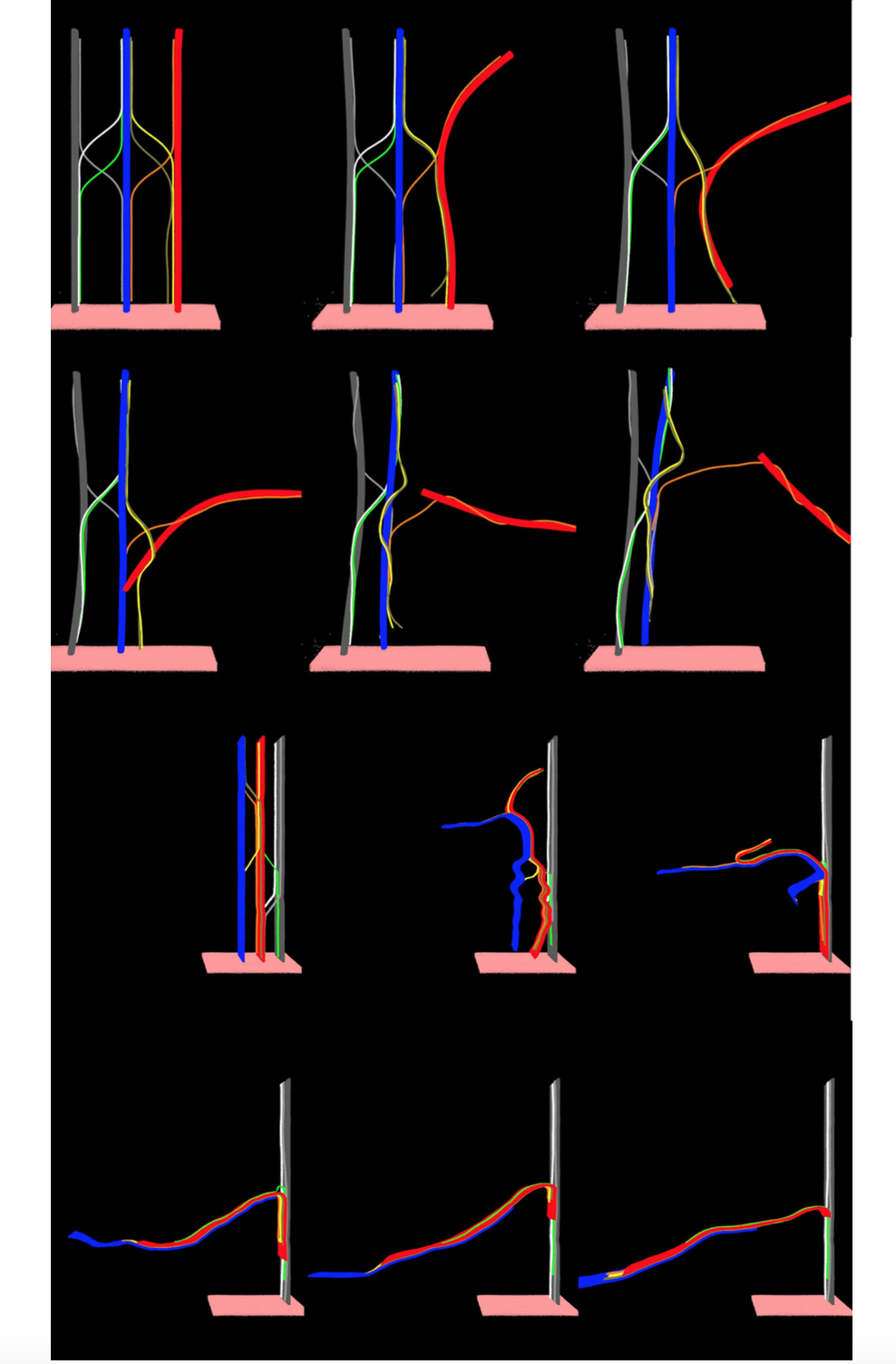
Machado, Leonardo D; Legoas, Sergio B; Galvao, Douglas S
Multi-Million Fully Atomistic Molecular Dynamics Simulations of Yarn Formation from Carbon Nanotube Forests Proceedings
Cambridge University Press, vol. 1407, 2012.
Abstract | Links | BibTeX | Tags: Carbon Nanotube Forests, Carbon Nanotubes, Molecular Dynamics, Yarns
@proceedings{machado2012multi,
title = {Multi-Million Fully Atomistic Molecular Dynamics Simulations of Yarn Formation from Carbon Nanotube Forests},
author = {Machado, Leonardo D and Legoas, Sergio B and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8537115&fulltextType=RA&fileId=S1946427412007105},
year = {2012},
date = {2012-01-01},
journal = {MRS Proceedings},
volume = {1407},
pages = {mrsf11--1407},
publisher = {Cambridge University Press},
abstract = {In this work we present preliminary results from multi-million fully atomistic classical molecular dynamics simulations carried out to test different existing mechanisms that have been proposed in the literature to explain the drawing of yarns from carbon nanotube forests. Despite the fact that it has been almost ten years since yarns were first drawn, there are still controversies on the mechanisms and necessary conditions that can produce yarns and sheets drawn from carbon nanotube forests. Moreover, few works have tried to understand at atomistic level the details of yarn drawing mechanisms, and no fully atomistic simulations have been carried out so far on this particular subject. Our preliminary results suggest that only direct van der Waals interactions among large bundles seem not to be enough to explain the yarn drawing process. Bundle interconnectors (such as small bundles connecting large bundles) were observed to play a critical role in our simulations. Depending on the topology of these interconnectors it was possible to observe from the simulations fibers/yarn formation from proposed structural models. These models were built based on structural information inferred from scanning electron microscopy data.},
keywords = {Carbon Nanotube Forests, Carbon Nanotubes, Molecular Dynamics, Yarns},
pubstate = {published},
tppubtype = {proceedings}
}

Legoas, SB; dos Santos, RPB; Troche, KS; Coluci, VR; Galvao, Douglas S
On the Existence of Ordered Phases of Encapsulated Diamondoids into Carbon Nanotubes Proceedings
Cambridge University Press, vol. 1407, 2012.
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, Diamondoids, Encapsulation
@proceedings{legoas2012existence,
title = {On the Existence of Ordered Phases of Encapsulated Diamondoids into Carbon Nanotubes},
author = {Legoas, SB and dos Santos, RPB and Troche, KS and Coluci, VR and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8539583&fileId=S194642741200704X},
year = {2012},
date = {2012-01-01},
journal = {MRS Proceedings},
volume = {1407},
pages = {mrsf11--1407},
publisher = {Cambridge University Press},
abstract = {We have investigated some diamondoids encapsulation into single walled carbon nanotubes (with diameters ranging from1.0 up to 2.2 nm) using fully atomistic molecular dynamics simulations. Diamondoids are the smallest hydrogen-terminated nanosized diamond-like molecules. Diamondois have been investigated for a large class of applications, ranging from oil industry to pharmaceuticals. Molecular ordered phases were observed for the encapsulation of adamantane, diamantane, and dihydroxy diamantanes. Chiral ordered phases, such as; double, triple, 4- and 5-stranded helices were also observed for those diamondoids. Our results also indicate that the modification of diamondoids through chemical functionalization with hydroxyl groups can lead to an enhancement of the molecular packing inside the carbon nanotubes in comparison to non-functionalized molecules. For larger diamondoids (such as, adamantane tetramers), we have not observed long-range ordering, but only a tendency of incomplete helical structural formation.},
keywords = {Carbon Nanotubes, Diamondoids, Encapsulation},
pubstate = {published},
tppubtype = {proceedings}
}
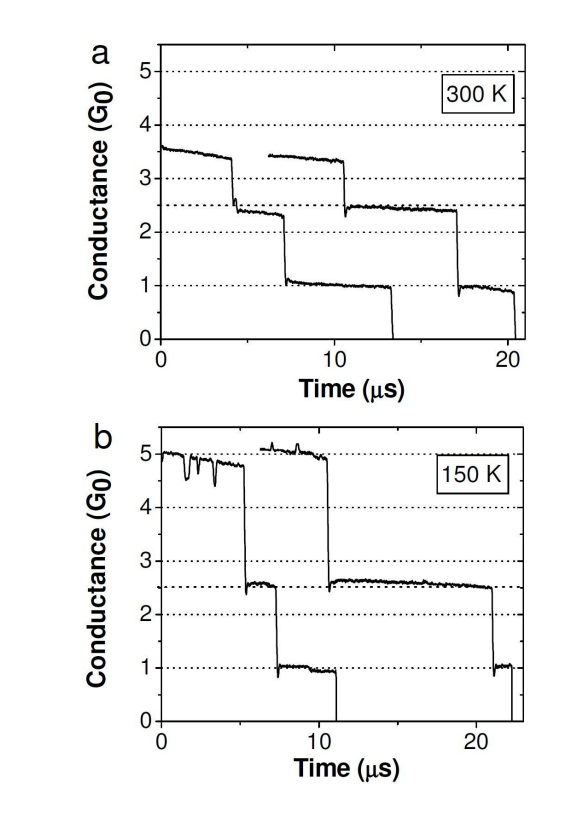
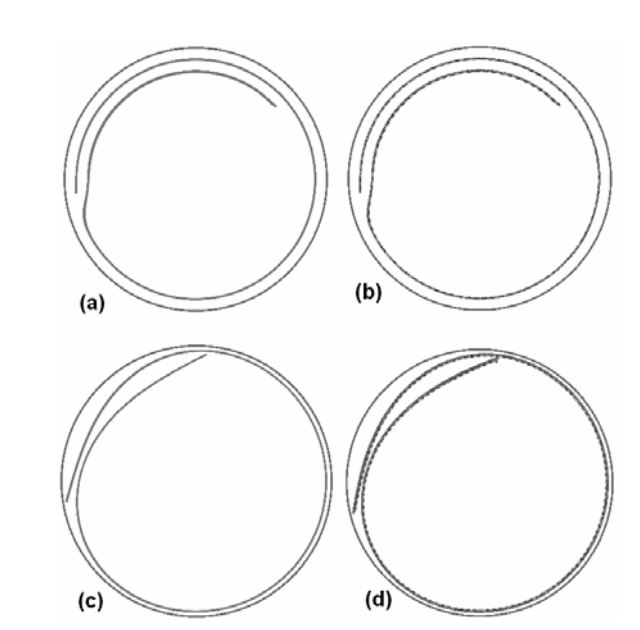
Lagos, MJ; Autreto, PAS; Galvao, DS; Ugarte, D
Correlation between Quantum Conductance and Atomic Arrangement of Silver Atomic-Size Nanowires Journal Article
In: arXiv preprint arXiv:1206.2551, 2012, (Draft version of: Correlation between quantum conductance and atomic arrangement of atomic-size silver nanowires Journal of Applied Physics, 111 (12), pp. 124316, 2012.).
Abstract | Links | BibTeX | Tags: Metallic Nanowires, Quantum Transport, TEM
@article{lagos2012correlation,
title = {Correlation between Quantum Conductance and Atomic Arrangement of Silver Atomic-Size Nanowires},
author = {Lagos, MJ and Autreto, PAS and Galvao, DS and Ugarte, D},
url = {http://arxiv.org/abs/1206.2551},
year = {2012},
date = {2012-01-01},
journal = {arXiv preprint arXiv:1206.2551},
abstract = {We have studied the effect of thermal effects on the structural and transport response of Ag atomic-size nanowires generated by mechanical elongation. Our study involves both time-resolved atomic resolution transmission electron microscopy imaging and quantum conductance measurement using an ultra-high-vacuum mechanically controllable break junction. We have observed drastic changes in conductance and structural properties of Ag nanowires generated at different temperatures (150 and 300 K). By combining electron microscopy images, electronic transport measurements and quantum transport calculations, we have been able to obtain a consistent correlation between the conductance and structural properties of Ag NWs. In particular, our study has revealed the formation of metastable rectangular rod-like Ag wire (3/3) along the (001) crystallographic direction, whose formation is enhanced. These results illustrate the high complexity of analyzing structural and quantum conductance behaviour of metal atomic-size wires; also, they reveal that it is extremely difficult to compare NW conductance experiments performed at different temperatures due to the fundamental modifications of the mechanical behavior.},
note = {Draft version of:
Correlation between quantum conductance and atomic arrangement of atomic-size silver nanowires
Journal of Applied Physics, 111 (12), pp. 124316, 2012.},
keywords = {Metallic Nanowires, Quantum Transport, TEM},
pubstate = {published},
tppubtype = {article}
}

Lagos, MJ; Autreto, PAS; Galvao, DS; Ugarte, D
Correlation between quantum conductance and atomic arrangement of atomic-size silver nanowires Journal Article
In: Journal of Applied Physics, vol. 111, no. 12, pp. 124316, 2012.
Abstract | Links | BibTeX | Tags: Metallic Nanowires, Quantum Transport, TEM
@article{lagos2012correlationb,
title = {Correlation between quantum conductance and atomic arrangement of atomic-size silver nanowires},
author = {Lagos, MJ and Autreto, PAS and Galvao, DS and Ugarte, D},
url = {http://scitation.aip.org/content/aip/journal/jap/111/12/10.1063/1.4729805},
year = {2012},
date = {2012-01-01},
journal = {Journal of Applied Physics},
volume = {111},
number = {12},
pages = {124316},
publisher = {AIP Publishing},
abstract = {We have studied the effect of thermal effects on the structural and transport response of Ag atomic-size nanowires (NWs) generated by mechanical elongation. Our study involves both time-resolved atomic resolution transmission electron microscopy imaging and quantum conductance measurement using an ultra-high-vacuum mechanically controllable break junction. We have observed drastic changes in conductance and structuralproperties of Agnanowires generated at different temperatures (150 and 300 K). By combining electron microscopy images, electronic transport measurements, and quantum transport calculations, we have been able to obtain a consistent correlation between the conductance and structuralproperties of Ag NWs. In particular, our study has revealed the formation of metastable rectangular rod-like Agwire (3/3) along the [001] crystallographic direction, whose formation is enhanced. These results illustrate the high complexity of analyzing structural and quantum conductance behaviour of metal atomic-size wires; also, they reveal that it is extremely difficult to compare NW conductance experiments performed at different temperatures due to the fundamental modifications of the mechanical behavior.
},
keywords = {Metallic Nanowires, Quantum Transport, TEM},
pubstate = {published},
tppubtype = {article}
}
Perim, Eric; Fonseca, Alexandre F; Galvao, Douglas S
When Small is Different: The Case of Membranes Inside Tubes Proceedings
Cambridge University Press, vol. 1451, 2012.
Abstract | Links | BibTeX | Tags: Mechanical Properties, Membranes, Nanoscale Effects, Scrolls
@proceedings{perim2012small,
title = {When Small is Different: The Case of Membranes Inside Tubes},
author = {Perim, Eric and Fonseca, Alexandre F and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8637821&fileId=S1946427412012523},
year = {2012},
date = {2012-01-01},
journal = {MRS Proceedings},
volume = {1451},
pages = {15--20},
publisher = {Cambridge University Press},
abstract = {Recently, classical elasticity theory for thin sheets was used to demonstrate the existence of a universal structural behavior describing the confinement of sheets inside cylindrical tubes. However, this kind of formalism was derived to describe macroscopic systems. A natural question is whether this behavior still holds at nanoscale. In this work, we have investigated through molecular dynamics simulations the structural behavior of graphene and boron nitride single layers confined into nanotubes. Our results show that the class of universality observed at macroscale is no longer observed at nanoscale. The origin of this discrepancy is addressed in terms of the relative importance of forces and energies at macro and nano scales.},
keywords = {Mechanical Properties, Membranes, Nanoscale Effects, Scrolls},
pubstate = {published},
tppubtype = {proceedings}
}

dos Santos, Ricardo P; Autreto, Pedro A; Perim, Eric; Brunetto, Gustavo; Galvao, Douglas S
On the Unzipping Mechanisms of Carbon Nanotubes: Insights from Reactive Molecular Dynamics Simulations Proceedings
Cambridge University Press, vol. 1451, 2012.
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, Molecular Dynamics, Unzipping
@proceedings{dos2012unzipping,
title = {On the Unzipping Mechanisms of Carbon Nanotubes: Insights from Reactive Molecular Dynamics Simulations},
author = {dos Santos, Ricardo P and Autreto, Pedro A and Perim, Eric and Brunetto, Gustavo and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8652294&fileId=S1946427412013292},
year = {2012},
date = {2012-01-01},
journal = {MRS Proceedings},
volume = {1451},
pages = {3--8},
publisher = {Cambridge University Press},
abstract = {Unzipping carbon nanotubes (CNTs) is considered one of the most promising approaches for the controlled and large-scale production of graphene nanoribbons (GNR). These structures are considered of great importance for the development of nanoelectronics because of its dimensions and intrinsic nonzero band gap value. Despite many years of investigations some details on the dynamics of the CNT fracture/unzipping processes remain unclear. In this work we have investigated some of these process through molecular dynamics simulations using reactive force fields (ReaxFF), as implemented in the Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) code. We considered multi-walled CNTs of different dimensions and chiralities and under induced mechanical stretching. Our preliminary results show that the unzipping mechanisms are highly dependent on CNT chirality. Well-defined and distinct fracture patterns were observed for the different chiralities. Armchair CNTs favor the creation of GNRs with well-defined armchair edges, while zigzag and chiral ones produce GNRs with less defined and defective edges.},
keywords = {Carbon Nanotubes, Molecular Dynamics, Unzipping},
pubstate = {published},
tppubtype = {proceedings}
}

Dos Santos, RPB; Perim, E; Autreto, PAS; Brunetto, Gustavo; Galvao, DS
On the unzipping of multiwalled carbon nanotubes Journal Article
In: Nanotechnology, vol. 23, no. 46, pp. 465702, 2012.
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, Fracture, Molecular Dynamics, Unzipping
@article{dos2012unzippingb,
title = {On the unzipping of multiwalled carbon nanotubes},
author = {Dos Santos, RPB and Perim, E and Autreto, PAS and Brunetto, Gustavo and Galvao, DS},
url = {http://iopscience.iop.org/0957-4484/23/46/465702},
year = {2012},
date = {2012-01-01},
journal = {Nanotechnology},
volume = {23},
number = {46},
pages = {465702},
publisher = {IOP Publishing},
abstract = {Graphene nanoribbons (GNRs) are very interesting structures which can retain graphene's high carrier mobility while presenting a finite bandgap. These properties make GNRs very valuable materials for the building of nanodevices. Unzipping carbon nanotubes (CNTs) is considered one of the most promising approaches for GNR controlled and large-scale production, although some of the details of the CNT unzipping processes are not completely known. In this work we have investigated CNT unzipping processes through fully atomistic molecular dynamics simulations using reactive force fields (ReaxFF). Multiwalled CNTs of different dimensions and chiralities under induced mechanical stretching were considered. Our results show that fracture patterns and stress profiles are highly CNT chirality dependent. Our results also show that the 'crests' (partially unzipped CNT regions presenting high curvature), originating from defective CNT areas, can act as a guide for the unzipping processes, which can explain the almost perfectly linear cuts frequently observed in unzipped CNTs.
},
keywords = {Carbon Nanotubes, Fracture, Molecular Dynamics, Unzipping},
pubstate = {published},
tppubtype = {article}
}

Lima, Marcio D; Li, Na; De Andrade, Monica Jung; Fang, Shaoli; Oh, Jiyoung; Spinks, Geoffrey M; Kozlov, Mikhail E; Haines, Carter S; Suh, Dongseok; Foroughi, Javad; Kim, Seon Jeong; Chen, Yongsheng; Ware, Taylor; Shin, Min Kyoon; Machado, Leonardo D; Fonseca, Alexandre F; Madden, John DW; Voit, Walter E; Galvao, Douglas S; Baughman, Ray H
Electrically, chemically, and photonically powered torsional and tensile actuation of hybrid carbon nanotube yarn muscles Journal Article
In: Science, vol. 338, no. 6109, pp. 928–932, 2012.
Abstract | Links | BibTeX | Tags: Actuation, Artificial Muscles, Carbon Nanotubes, top20, Yarns
@article{lima2012electrically,
title = {Electrically, chemically, and photonically powered torsional and tensile actuation of hybrid carbon nanotube yarn muscles},
author = {Lima, Marcio D and Li, Na and De Andrade, Monica Jung and Fang, Shaoli and Oh, Jiyoung and Spinks, Geoffrey M and Kozlov, Mikhail E and Haines, Carter S and Suh, Dongseok and Foroughi, Javad and Kim, Seon Jeong and Chen, Yongsheng and Ware, Taylor and Shin, Min Kyoon and Machado, Leonardo D and Fonseca, Alexandre F and Madden, John DW and Voit, Walter E and Galvao, Douglas S and Baughman, Ray H
},
url = {http://www.sciencemag.org/content/338/6109/928.short},
year = {2012},
date = {2012-01-01},
journal = {Science},
volume = {338},
number = {6109},
pages = {928--932},
publisher = {American Association for the Advancement of Science},
abstract = {Artificial muscles are of practical interest, but few types have been commercially exploited. Typical problems include slow response, low strain and force generation, short cycle life, use of electrolytes, and low energy efficiency. We have designed guest-filled, twist-spun carbon nanotube yarns as electrolyte-free muscles that provide fast, high-force, large-stroke torsional and tensile actuation. More than a million torsional and tensile actuation cycles are demonstrated, wherein a muscle spins a rotor at an average 11,500 revolutions/minute or delivers 3% tensile contraction at 1200 cycles/minute. Electrical, chemical, or photonic excitation of hybrid yarns changes guest dimensions and generates torsional rotation and contraction of the yarn host. Demonstrations include torsional motors, contractile muscles, and sensors that capture the energy of the sensing process to mechanically actuate.},
keywords = {Actuation, Artificial Muscles, Carbon Nanotubes, top20, Yarns},
pubstate = {published},
tppubtype = {article}
}
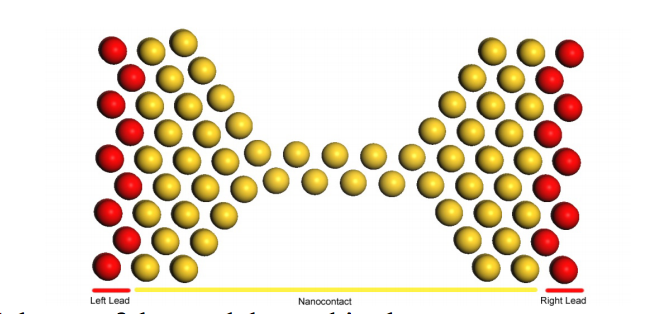
Autreto, Pedro A; Lagos, Maureen J; Ugarte, Daniel; Galvao, Douglas S
Correlation Between Quantum Conductance and Atomic Arrangement of Silver Atomic-Size Nanocontacts Proceedings
Cambridge University Press, vol. 1429, 2012.
Abstract | Links | BibTeX | Tags: Metallic Nanowires, Quantum Transport, TEM
@proceedings{autreto2012correlation,
title = {Correlation Between Quantum Conductance and Atomic Arrangement of Silver Atomic-Size Nanocontacts},
author = {Autreto, Pedro A and Lagos, Maureen J and Ugarte, Daniel and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8717061&fileId=S1946427412015011},
year = {2012},
date = {2012-01-01},
journal = {MRS Proceedings},
volume = {1429},
pages = {mrss12--1429},
publisher = {Cambridge University Press},
abstract = {In this work we have studied the importance of thermal effects on the structural and transport properties of Ag atomic-size nanowires (NWs) generated by mechanical stretching. Our study involve time-resolved atomic high resolution transmission electron microscopy imaging and quantum conductance measurement using an ultra-high-vacuum mechanically controllable break junction combined with quantum transport calculations. We have observed drastic changes in conductance and structural properties of Ag NWs generated at different temperatures (150 and 300 K). By combining electron microscopy images, electronic transport measurements and theoretical modeling, we have been able to establish a consistent correlation between the conductance and structural properties of Ag NWs. In particular, our study has revealed the formation of metastable rectangular rod-like Ag wires along the [001] crystallographic direction.},
keywords = {Metallic Nanowires, Quantum Transport, TEM},
pubstate = {published},
tppubtype = {proceedings}
}

dos Santos, Ricardo P; Machado, Leonardo D; Legoas, Sergio B; Galvao, Douglas S
Tribological Properties of Graphene and Boron-Nitride Layers: A Fully Atomistic Molecular Dynamics Study Proceedings
Cambridge University Press, vol. 1407, 2012.
Abstract | Links | BibTeX | Tags: Boron Nitride, Graphene, Molecular Dynamics, Tribology
@proceedings{dos2012tribological,
title = {Tribological Properties of Graphene and Boron-Nitride Layers: A Fully Atomistic Molecular Dynamics Study},
author = {dos Santos, Ricardo P and Machado, Leonardo D and Legoas, Sergio B and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8537106&fileId=S1946427412007063},
year = {2012},
date = {2012-01-01},
journal = {MRS Proceedings},
volume = {1407},
pages = {mrsf11--1407},
publisher = {Cambridge University Press},
abstract = {Graphene has been one of the most important subjects in materials science in the last years. Recently, the frictional characteristics of atomically thin sheets were experimentally investigated using atomic force microscopy (AFM). A new mechanism to explain the enhanced friction for these materials, based on elastic compliance has been proposed. Here, we have investigated the tribological properties of graphene and boron-nitride (single and multi-layers) membranes using fully atomistic molecular dynamics simulations. These simulations were carried out using classical force fields, as implemented in the Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) code. The used structural models contain typically hundreds of thousands of atoms. In order to mimic the experimental conditions, an artificial AFM tip was moved over the membranes and the tribological characteristics determined in terms of forces and energies. Our results are in good agreement with the available experimental data. They show that the observed enhanced tribological properties can be explained in terms of out-of-plane geometrical distortions and elastic waves propagation. They validate the general features of the model proposed by Lee et al. (Science 328, 76 (2010).},
keywords = {Boron Nitride, Graphene, Molecular Dynamics, Tribology},
pubstate = {published},
tppubtype = {proceedings}
}
2011
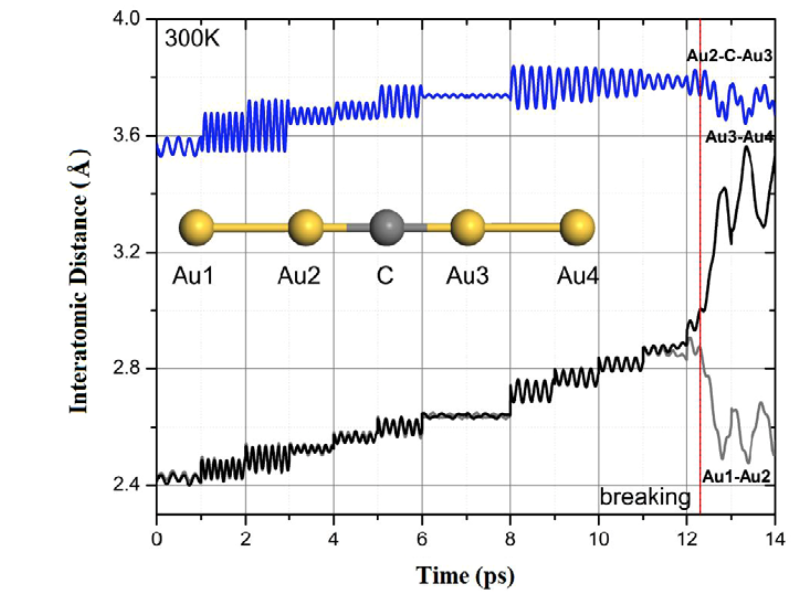
PAS Autreto MJ Lagos, SB Legoas
Temperature effects on the occurrence of long interatomic distances in atomic chains formed from stretched gold nanowires Journal Article
In: Nanotechnology, vol. 22, no. 9, pp. 095705, 2011.
Abstract | Links | BibTeX | Tags: DFT, Gold, Metallic Nanowires, TEM
@article{Lagos2011,
title = {Temperature effects on the occurrence of long interatomic distances in atomic chains formed from stretched gold nanowires},
author = {MJ Lagos, PAS Autreto, SB Legoas, F Sato, V Rodrigues, DS Galvao, D Ugarte},
url = {http://iopscience.iop.org/0957-4484/22/9/095705},
year = {2011},
date = {2011-03-04},
journal = {Nanotechnology},
volume = {22},
number = {9},
pages = {095705},
abstract = {The origin of long interatomic distances in suspended gold atomic chains formed from stretched nanowires remains the object of debate despite the large amount of theoretical and experimental work. Here, we report new atomic resolution electron microscopy observations acquired at room and liquid-nitrogen temperatures and theoretical results from ab initio quantum molecular dynamics on chain formation and stability. These new data are suggestive that the long distances are due to contamination by carbon atoms originating from the decomposition of adsorbed hydrocarbon molecules.
},
keywords = {DFT, Gold, Metallic Nanowires, TEM},
pubstate = {published},
tppubtype = {article}
}
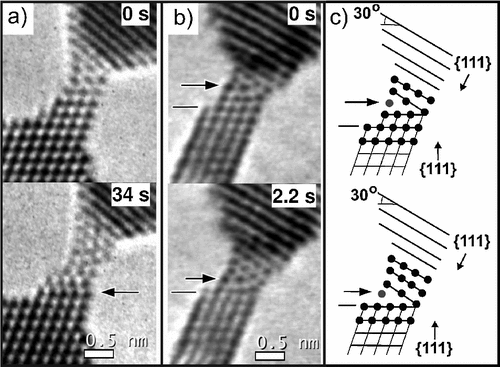
Lagos, Maureen J; Sato, Fernando; Galvao, Douglas S; Ugarte, Daniel
Mechanical deformation of nanoscale metal rods: when size and shape matter Journal Article
In: Physical Review Letters, vol. 106, no. 5, pp. 055501, 2011.
Abstract | Links | BibTeX | Tags: Defects, DFT, Mechanical Properties, Metallic Nanowires
@article{lagos2011mechanical,
title = {Mechanical deformation of nanoscale metal rods: when size and shape matter},
author = {Lagos, Maureen J and Sato, Fernando and Galvao, Douglas S and Ugarte, Daniel},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.106.055501},
year = {2011},
date = {2011-01-01},
journal = {Physical Review Letters},
volume = {106},
number = {5},
pages = {055501},
publisher = {American Physical Society},
abstract = {Face centered cubic metals deform mainly by propagating partial dislocations generating planar fault ribbons. How do metals deform if the size is smaller than the fault ribbons? We studied the elongation of Au and Pt nanorods by in situ electron microscopy and ab initio calculations. Planar fault activation barriers are so low that, for each temperature, a minimal rod size is required to become active for releasing elastic energy. Surface effects dominate deformation energetics; system size and shape determine the preferred fault gliding directions which induce different tensile and compressive behavior.
},
keywords = {Defects, DFT, Mechanical Properties, Metallic Nanowires},
pubstate = {published},
tppubtype = {article}
}

Machado, Leonardo D; Legoas, Sergio B; Soares, Jaqueline S; Shadmi, Nitzan; Jorio, Ado; Joselevich, Ernesto; Galvao, Douglas S
Cambridge University Press, vol. 1284, 2011.
Abstract | Links | BibTeX | Tags: Mechanical Properties, Molecular Dynamics, Serpentines
@proceedings{machado2011formation,
title = {On the formation of carbon nanotube serpentines: insights from multi-million atom molecular dynamics simulation},
author = {Machado, Leonardo D and Legoas, Sergio B and Soares, Jaqueline S and Shadmi, Nitzan and Jorio, Ado and Joselevich, Ernesto and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8194288&fileId=S194642741100220X},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1284},
pages = {mrsf10--1284},
publisher = {Cambridge University Press},
abstract = {In this work we present preliminary results from molecular dynamics simulations for carbon nanotubes serpentine dynamics formation. These S-like nanostructures consist of a series of parallel and straight nanotube segments connected by alternating U-turn shaped curves. Nanotube serpentines were experimentally synthesized and reported in recent years, but up to now no atomistic simulations have been carried out to address the dynamics of formation of these structures. We have carried out fully atomistic molecular dynamics simulations in the framework of classical mechanics with a standard molecular force field. Multi-million atoms structures formed by stepped substrates with a carbon nanotube (about 1 micron in length) placed on top of them have been considered in our simulations. A force is applied to the upper part of the tube during a short period of time and then turned off and the system set free to evolve in time. Our results showed that these conditions are sufficient to form robust serpentines and validate the general features of the ‘falling spaghetti mechanism’ previously proposed to explain their formation.},
keywords = {Mechanical Properties, Molecular Dynamics, Serpentines},
pubstate = {published},
tppubtype = {proceedings}
}

Autreto, PAS; Lagos, MJ; Sato, F; Bettini, J; Rocha, AR; Rodrigues, V; Ugarte, D; Galvao, DS
Intrinsic Stability of the Smallest Possible Silver Nanotube Journal Article
In: Physical Review Letters, vol. 106, no. 6, pp. 065501, 2011.
Abstract | Links | BibTeX | Tags: DFT, Mechanical Properties, Metallic Nanowires, New Structures, top20
@article{autreto2011intrinsic,
title = {Intrinsic Stability of the Smallest Possible Silver Nanotube},
author = {Autreto, PAS and Lagos, MJ and Sato, F and Bettini, J and Rocha, AR and Rodrigues, V and Ugarte, D and Galvao, DS},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.106.065501},
year = {2011},
date = {2011-01-01},
journal = {Physical Review Letters},
volume = {106},
number = {6},
pages = {065501},
publisher = {American Physical Society},
abstract = {Recently, Lagos et al. [Nature Nanotech. 4, 149 (2009)] reported the discovery of the smallest possible Ag nanotube with a square cross section. Ab initio density functional theory calculations strongly support that the stability of these hollow structures is structurally intrinsic and not the result of contamination by light atoms. We also report the first experimental observation of the theoretically predicted corrugation of the hollow structure. Quantum conductance calculations predict a unique signature of 3.6G0 for this new family of nanotubes.},
keywords = {DFT, Mechanical Properties, Metallic Nanowires, New Structures, top20},
pubstate = {published},
tppubtype = {article}
}
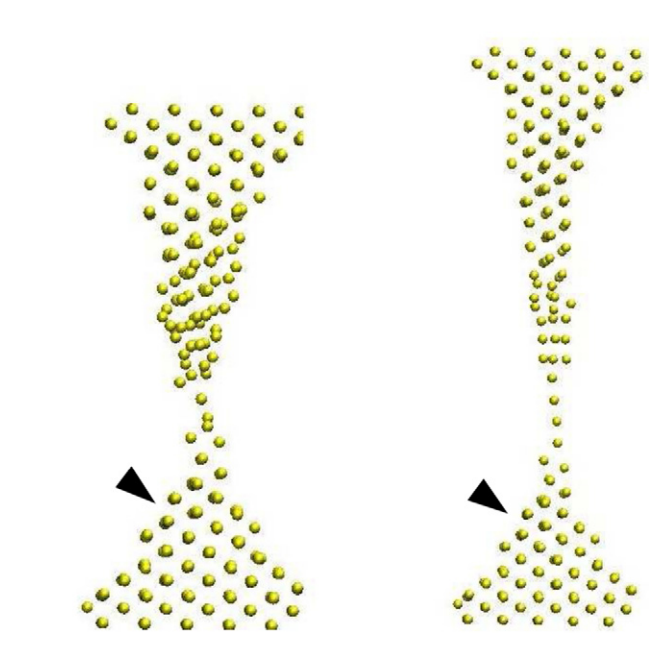
Lagos, MJ; Autreto, PAS; Legoas, SB; Sato, F; Rodrigues, V; Galvao, DS; Ugarte, D
Temperature effects on the occurrence of long interatomic distances in atomic chains formed from stretched gold nanowires Journal Article
In: Nanotechnology, vol. 22, no. 9, pp. 095705, 2011.
Abstract | Links | BibTeX | Tags: Gold, Mechanical Properties, Metallic Nanowires
@article{lagos2011temperature,
title = {Temperature effects on the occurrence of long interatomic distances in atomic chains formed from stretched gold nanowires},
author = {Lagos, MJ and Autreto, PAS and Legoas, SB and Sato, F and Rodrigues, V and Galvao, DS and Ugarte, D},
url = {http://iopscience.iop.org/0957-4484/21/48/485702},
year = {2011},
date = {2011-01-01},
journal = {Nanotechnology},
volume = {22},
number = {9},
pages = {095705},
publisher = {IOP Publishing},
abstract = {We have studied the changes induced by thermal effects in the structural and transport response of Au nanowires generated by mechanical elongation. We have used time-resolved atomic resolution transmission electron microscopy imaging and quantum conductance measurement using a mechanically controllable break junction. Our results showed remarkable differences in the NW evolution for experiments realized at 150 and 300 K, which modifies drastically the conductance response during elongation. Molecular dynamics and electronic transport calculations were used to consistently correlate the observed structural and conductance behavior. These results emphasize that it is essential to take into account the precise atomic arrangement of nanocontacts generated by mechanical stretching to understand electrical transport properties. Also, our study shows that much care must be taken when comparing results obtained in different experimental conditions, mainly different temperatures.
},
keywords = {Gold, Mechanical Properties, Metallic Nanowires},
pubstate = {published},
tppubtype = {article}
}
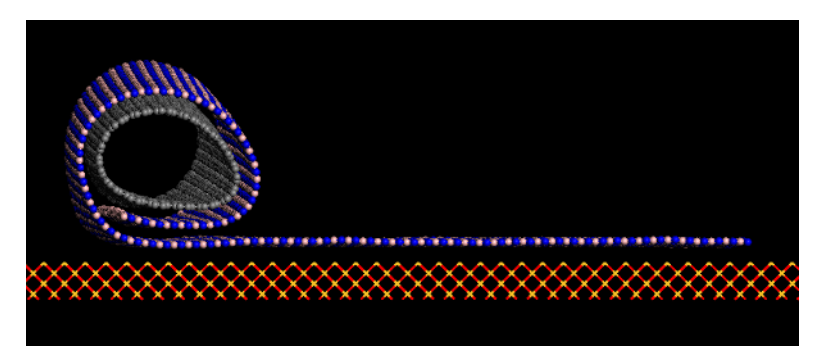
Perim, Eric; Galvao, Douglas S
Stability and Dynamics of Boron Nitride Nanoscrolls Proceedings
Cambridge University Press, vol. 1307, 2011.
Abstract | Links | BibTeX | Tags: Boron Nitride, Molecular Dynamics, Scrolls
@proceedings{perim2011stability,
title = {Stability and Dynamics of Boron Nitride Nanoscrolls},
author = {Perim, Eric and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayFulltext?type=1&fid=8200678&jid=OPL&volumeId=1307&issueId=-1&aid=8200676},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1307},
pages = {mrsf10--1307},
publisher = {Cambridge University Press},
abstract = {We report here molecular dynamics results for boron nitride nanoscroll structures
(BNNSs) with relation to their stability and formation mechanisms. We show that, similarly to
carbon nanoscrolls, BNNSs are stable due to van der Waals interactions among overlapping
layers. The energy balance between losses and gains (due to elastic deformations and van der
Waals interactions, respectively) when the structure is rolled up leads to the existence of a
critical value of the internal scroll diameter where stable or metastable structures can be formed.
The mechanisms of scroll formation and stability as a function of their chirality were also
investigated.},
keywords = {Boron Nitride, Molecular Dynamics, Scrolls},
pubstate = {published},
tppubtype = {proceedings}
}
(BNNSs) with relation to their stability and formation mechanisms. We show that, similarly to
carbon nanoscrolls, BNNSs are stable due to van der Waals interactions among overlapping
layers. The energy balance between losses and gains (due to elastic deformations and van der
Waals interactions, respectively) when the structure is rolled up leads to the existence of a
critical value of the internal scroll diameter where stable or metastable structures can be formed.
The mechanisms of scroll formation and stability as a function of their chirality were also
investigated.
Autreto, Pedro AS; Flores, Marcelo Z; Legoas, Sergio B; Santos, Ricardo PB; Galvao, Douglas S
Cambridge University Press, vol. 1284, 2011.
Abstract | Links | BibTeX | Tags: Graphane, Graphene, Hydrogenation, Molecular Dynamics
@proceedings{autreto2011fully,
title = {A Fully Atomistic Reactive Molecular Dynamics Study on the Formation of Graphane from Graphene Hydrogenated Membranes.},
author = {Autreto, Pedro AS and Flores, Marcelo Z and Legoas, Sergio B and Santos, Ricardo PB and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8364784&fileId=S1946427411013583},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1284},
pages = {mrsf10--1284},
publisher = {Cambridge University Press},
abstract = {Using fully reactive molecular dynamics methodologies we investigated the structural and dynamical aspects of the fluorination mechanism leading to fluorographene formation from graphene membranes. Fluorination tends to produce significant defective areas on the membranes with variation on the typical carbon-carbon distances, sometimes with the presence of large holes due to carbon losses. The results obtained in our simulations are in good agreement with the broad distribution of values for the lattice parameter experimentally observed. We have also investigated mixed atmospheres composed by H and F atoms. When H is present in small quantities an expressive reduction on the rate of incorporation of fluorine was observed. On the other hand when fluorine atoms are present in small quantities in a hydrogen atmosphere, they induce an increasing on the hydrogen incorporation and the formation of locally self-organized structure of adsorbed H and F atoms.},
keywords = {Graphane, Graphene, Hydrogenation, Molecular Dynamics},
pubstate = {published},
tppubtype = {proceedings}
}
Azevedo, David L; Sato, Fernando; Gomes de Sousa Filho, Antonio; Galvao, Douglas S
In: Molecular Simulation, vol. 37, no. 9, pp. 746–751, 2011.
Abstract | Links | BibTeX | Tags: CNT encapsulation, Cobaltocene, Molecular Dynamics
@article{azevedo2011van,
title = {van der Waals potential barrier for cobaltocene encapsulation into single-walled carbon nanotubes: classical molecular dynamics and ab initio study},
author = {Azevedo, David L and Sato, Fernando and Gomes de Sousa Filho, Antonio and Galvao, Douglas S},
url = {http://www.tandfonline.com/doi/abs/10.1080/08927022.2010.537093#.VLfBForF-2o},
year = {2011},
date = {2011-01-01},
journal = {Molecular Simulation},
volume = {37},
number = {9},
pages = {746--751},
publisher = {Taylor & Francis Group},
abstract = {In this work, we carried out geometry optimisations and classical molecular dynamics for the problem of cobaltocene (CC) encapsulation into different carbon nanotubes (CNTs) ((7,7), (8,8), (13,0) and (14,0) tubes were used). CCs are molecules composed of two aromatic pentagonal rings (C5H5) sandwiching one cobalt atom. From our simulation results, we observed that CC was encapsulated into CNTs (8,8), (13,0) and (14,0). However, for CNT (7,7), the encapsulation could not occur, in disaggrement with some previous works in the literature. Our results show that the encapsulation process is mainly governed by van der Waals potential barriers.},
keywords = {CNT encapsulation, Cobaltocene, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Santos, Ricardo PB; Autreto, Pedro AS; Legoas, Sergio B; Galvao, Douglas S
Cambridge University Press, vol. 1344, 2011.
Abstract | Links | BibTeX | Tags: Fluorographene, Functionalization, Graphane, Graphene
@proceedings{santos2011dynamics,
title = {The Dynamics of Formation of Graphane-like Fluorinated Graphene Membranes (Fluorographene): A Reactive Molecular Dynamics Study},
author = {Santos, Ricardo PB and Autreto, Pedro AS and Legoas, Sergio B and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayFulltext?type=1&fid=8237871&jid=OPL&volumeId=1284&issueId=-1&aid=8237869},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1344},
pages = {mrss11--1344},
publisher = {Cambridge University Press},
abstract = {Recently, Elias et al. (Science 323, 610 (2009).) reported the experimental realization of
the formation of graphane from hydrogenation of graphene membranes under cold plasma
exposure. In graphane, the carbon-carbon bonds are in sp3
configuration, as opposed to the sp2
hybridization of graphene, and the C–H bonds exhibit an alternating pattern (up and down with
relation to the plane defined by the carbon atoms). In this work we have investigated, using
reactive molecular dynamics simulations, the role of H frustration (breaking the H atoms up and
down alternating pattern) in graphane-like structures. Our results show that a significant
percentage of uncorrelated H frustrated domains are formed in the early stages of the
hydrogenation process, leading to membrane shrinkage and extensive membrane corrugations.
This might explain the significant broad distribution of values of lattice parameter
experimentally observed. For comparison purposes we have also analyzed fluorinated graphanelike
structures. Our results show that similarly to H, F atoms also create significant uncorrelated
frustrated domains on graphene membranes. },
keywords = {Fluorographene, Functionalization, Graphane, Graphene},
pubstate = {published},
tppubtype = {proceedings}
}
the formation of graphane from hydrogenation of graphene membranes under cold plasma
exposure. In graphane, the carbon-carbon bonds are in sp3
configuration, as opposed to the sp2
hybridization of graphene, and the C–H bonds exhibit an alternating pattern (up and down with
relation to the plane defined by the carbon atoms). In this work we have investigated, using
reactive molecular dynamics simulations, the role of H frustration (breaking the H atoms up and
down alternating pattern) in graphane-like structures. Our results show that a significant
percentage of uncorrelated H frustrated domains are formed in the early stages of the
hydrogenation process, leading to membrane shrinkage and extensive membrane corrugations.
This might explain the significant broad distribution of values of lattice parameter
experimentally observed. For comparison purposes we have also analyzed fluorinated graphanelike
structures. Our results show that similarly to H, F atoms also create significant uncorrelated
frustrated domains on graphene membranes.
Legoas, SB; Dos Santos, RPB; Troche, KS; Coluci, VR; Galvao, DS
Ordered phases of encapsulated diamondoids into carbon nanotubes Journal Article
In: Nanotechnology, vol. 22, no. 31, pp. 315708, 2011.
Abstract | Links | BibTeX | Tags: CNT encapsulation, Diamondoids, Molecular Dynamics
@article{legoas2011ordered,
title = {Ordered phases of encapsulated diamondoids into carbon nanotubes},
author = {Legoas, SB and Dos Santos, RPB and Troche, KS and Coluci, VR and Galvao, DS},
url = {http://iopscience.iop.org/0957-4484/22/31/315708},
year = {2011},
date = {2011-01-01},
journal = {Nanotechnology},
volume = {22},
number = {31},
pages = {315708},
publisher = {IOP Publishing},
abstract = {Diamondoids are hydrogen-terminated nanosized diamond fragments that are present in petroleum crude oil at low concentrations. These fragments are found as oligomers of the smallest diamondoid, adamantane (C10H16). Due to their small size, diamondoids can be encapsulated into carbon nanotubes to form linear arrangements. We have investigated the encapsulation of diamondoids into single walled carbon nanotubes with diameters between 1.0 and 2.2 nm using fully atomistic simulations. We performed classical molecular dynamics and energy minimizations calculations to determine the most stable configurations. We observed molecular ordered phases (e.g. double, triple, 4- and 5-stranded helices) for the encapsulation of adamantane, diamantane, and dihydroxy diamantane. Our results also indicate that the functionalization of diamantane with hydroxyl groups can lead to an improvement on the molecular packing factor when compared to non-functionalized compounds. Comparisons to hard-sphere models revealed differences, especially when more asymmetrical diamondoids were considered. For larger diamondoids (i.e., adamantane tetramers), we have not observed long-range ordering but only a tendency to form incomplete helical structures. Our calculations predict that thermally stable (at least up to room temperature) complex ordered phases of diamondoids can be formed through encapsulation into carbon nanotubes.},
keywords = {CNT encapsulation, Diamondoids, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Coutinho, Samir S; Azevedo, David L; Galvao, Douglas S
Tuning Electronic and Structural Properties of Triple Layers of Intercalated Graphene and Hexagonal Boron Nitride: An Ab-initio Study. Journal Article
In: MRS Proceedings, vol. 1307, pp. mrsf10–1307, 2011.
Abstract | Links | BibTeX | Tags: BN, DFT, Graphene, Heterostructures
@article{coutinho2011tuning,
title = {Tuning Electronic and Structural Properties of Triple Layers of Intercalated Graphene and Hexagonal Boron Nitride: An Ab-initio Study.},
author = {Coutinho, Samir S and Azevedo, David L and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8330317&fileId=S1946427411003642},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1307},
pages = {mrsf10--1307},
publisher = {Cambridge University Press},
abstract = {Recently, several experiments and theoretical studies demonstrated the possibility of tuning or modulating band gap values of nanostructures composed of bi-layer graphene, bi-layer hexagonal boron-nitride (BN) and hetero-layer combinations. These triple layers systems present several possibilities of stacking. In this work we report an ab initio (within the formalism of density functional theory (DFT)) study of structural and electronic properties of some of these stacked configurations. We observe that an applied external electric field can alter the electronic and structural properties of these systems. With the same value of the applied electric field the band gap values can be increased or decreased, depending on the layer stacking sequences. Strong geometrical deformations were observed. These results show that the application of an external electric field perpendicular to the stacked layers can effectively be used to modulate their inter-layer distances and/or their band gap values.},
keywords = {BN, DFT, Graphene, Heterostructures},
pubstate = {published},
tppubtype = {article}
}
Brunetto, Gustavo; Legoas, Sergio B; Coluci, Vitor R; Lucena, Liacir S; Galvao, Douglas S
Dynamics of Graphene Nanodrums Proceedings
Cambridge University Press, vol. 1284, 2011.
Abstract | Links | BibTeX | Tags: Graphene Membranes, Mechanical Properties, Nanodrum
@proceedings{brunetto2011dynamics,
title = {Dynamics of Graphene Nanodrums},
author = {Brunetto, Gustavo and Legoas, Sergio B and Coluci, Vitor R and Lucena, Liacir S and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8195889&fileId=S1946427411002272},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1284},
pages = {mrsf10--1284},
publisher = {Cambridge University Press},
abstract = {Recently, it was proposed that graphene sheets deposited on silicon oxide can act as impermeable atomic membranes to standard gases, such as helium, argon, and nitrogen. It is assumed that graphene membrane is clamped over the surface due only to van der Waals forces. The leakage mechanism can be experimentally addressed only indirectly. In this work we have carried out molecular dynamics simulations to study this problem. We have considered nano-containers composed of a chamber of silicon oxide filled with gas and sealed by single and multi-layer graphene membranes. The obtained results are in good qualitative agreement with the experimental data. We observed that the graphene membranes remain attached to the substrate for pressure values up to two times the largest value experimentally investigated. We did not observe any gas leakage through the membrane/substrate interface until the critical limit is reached and then a sudden membrane detachment occurs.},
keywords = {Graphene Membranes, Mechanical Properties, Nanodrum},
pubstate = {published},
tppubtype = {proceedings}
}
Vasconcelos, MS; Azevedo, David L; Hadad, A; Galvao, DS
Electronic properties of Fibonacci and random Si--Ge chains Journal Article
In: Journal of Physics: Condensed Matter, vol. 23, no. 40, pp. 405501, 2011.
Abstract | Links | BibTeX | Tags: Electronic Structure, Fibonacci, Si-Ge chains
@article{vasconcelos2011electronic,
title = {Electronic properties of Fibonacci and random Si--Ge chains},
author = {Vasconcelos, MS and Azevedo, David L and Hadad, A and Galvao, DS},
url = {http://iopscience.iop.org/0953-8984/23/40/405501},
year = {2011},
date = {2011-01-01},
journal = {Journal of Physics: Condensed Matter},
volume = {23},
number = {40},
pages = {405501},
publisher = {IOP Publishing},
abstract = {In this paper we address a theoretical calculation of the electronic spectra of an Si–Ge atomic chain that is arranged in a Fibonacci quasi-periodic sequence, by using a semi-empirical quantum method based on the Hückel extended model. We apply the Fibonacci substitutional sequences in the atomic building blocks A(Si) and B(Ge) through the inflation rule or a recursion relation. In our ab initio calculations we use only a single point, which is sufficient for considering all the orbitals and charge distribution across the entire system. Although the calculations presented here are more complete than the models adopted in the literature which take into account the electronic interaction only up to the second and third neighbors, an interesting property remains in their electronic spectra: the fractality (which is the main signature of this kind of system). We discuss this fractality of the spectra and we compare them with the random arrangement of the Si–Ge atomic chain, and with previous results based on the tight-binding approximation of the Schrödinger equation considering up to the nearest neighbor.
},
keywords = {Electronic Structure, Fibonacci, Si-Ge chains},
pubstate = {published},
tppubtype = {article}
}
Brunetto, Gustavo; Sato, Fernando; Bouju, Xavier; Galvao, Douglas S
The First Molecular Wheel: A Theoretical Investigation Proceedings
Cambridge University Press, vol. 1286, 2011.
Abstract | Links | BibTeX | Tags: Molecular Dynamics, Molecular Electronics, Nanowheel
@proceedings{brunetto2011first,
title = {The First Molecular Wheel: A Theoretical Investigation},
author = {Brunetto, Gustavo and Sato, Fernando and Bouju, Xavier and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=7975705&fileId=S1946427411000133},
year = {2011},
date = {2011-01-01},
journal = {MRS Proceedings},
volume = {1286},
pages = {mrsf10--1286},
publisher = {Cambridge University Press},
abstract = {Recently, the first molecular nanowheel was synthesized and characterized from Scanning Tunneling Microscope (STM) experiments. It was demonstrated that a specifically designed hydrocarbon molecule (C44H24) could roll on a copper substrate along the [110] surface direction. In this work we report a preliminary theoretical analysis of the isolated molecule and of its rolling processes on different Cu surfaces. We have used ab initio and classical molecular dynamics methods. The simulations showed that the rolling mechanism is only possible for the [110] surface. In this case, the spatial separation among rows of copper atoms is enough to ‘trap’ the molecule and to create the necessary torque to roll it. Other surface orientations ([111] and [100]) are too smooth and cannot provide the necessary torque for the rolling process.},
keywords = {Molecular Dynamics, Molecular Electronics, Nanowheel},
pubstate = {published},
tppubtype = {proceedings}
}
2010
Martins, BVC; Galvao, DS
Curved graphene nanoribbons: structure and dynamics of carbon nanobelts Journal Article
In: Nanotechnology, vol. 21, no. 7, pp. 075710, 2010.
Abstract | Links | BibTeX | Tags: Folding, Graphene, Mechanical Properties, Nanobelts, NanoRibbons
@article{martins2010curved,
title = {Curved graphene nanoribbons: structure and dynamics of carbon nanobelts},
author = {Martins, BVC and Galvao, DS},
url = {http://iopscience.iop.org/0957-4484/21/7/075710},
year = {2010},
date = {2010-01-01},
journal = {Nanotechnology},
volume = {21},
number = {7},
pages = {075710},
publisher = {IOP Publishing},
abstract = {Carbon nanoribbons (CNRs) are graphene (planar) structures with a large aspect ratio. Carbon nanobelts (CNBs) are small graphene nanoribbons rolled up into spiral-like structures, i.e. carbon nanoscrolls (CNSs) with a large aspect ratio. In this work we investigated the energetics and dynamical aspects of CNBs formed from rolling up CNRs. We have carried out molecular dynamics simulations using reactive empirical bond-order potentials. Our results show that, similarly to CNSs, CNB formation is dominated by two major energy contributions, the increase in the elastic energy due to the bending of the initial planar configuration (decreasing structural stability) and the energetic gain due to van der Waals interactions of the overlapping surface of the rolled layers (increasing structural stability). Beyond a critical diameter value these scrolled structures can be even more stable (in terms of energy) than their equivalent planar configurations. In contrast to CNSs that require energy-assisted processes (sonication, chemical reactions, etc) to be formed, CNBs can be spontaneously formed from low temperature driven processes. Long CNBs (length of ~30.0 nm) tend to exhibit self-folded racket-like conformations with formation dynamics very similar to the one observed for long carbon nanotubes. Shorter CNBs will be more likely to form perfect scrolled structures. Possible synthetic routes to fabricate CNBs from graphene membranes are also addressed.
},
keywords = {Folding, Graphene, Mechanical Properties, Nanobelts, NanoRibbons},
pubstate = {published},
tppubtype = {article}
}
Autreto, PAS; Legoas, SB; Flores, MZS; Galvao, DS
Carbon nanotube with square cross-section: An ab initio investigation Journal Article
In: The Journal of chemical physics, vol. 133, no. 12, pp. 124513, 2010.
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, DFT, New Structures, square tubes
@article{autreto2010carbon,
title = {Carbon nanotube with square cross-section: An ab initio investigation},
author = {Autreto, PAS and Legoas, SB and Flores, MZS and Galvao, DS},
url = {http://scitation.aip.org/content/aip/journal/jcp/133/12/10.1063/1.3483237},
year = {2010},
date = {2010-01-01},
journal = {The Journal of chemical physics},
volume = {133},
number = {12},
pages = {124513},
publisher = {AIP Publishing},
abstract = {Recently, Lagos et al. [Nat. Nanotechnol.4, 149 (2009)] reported the discovery of the smallest possible silver square cross-section nanotube. A natural question is whether similar carbon nanotubes can exist. In this work we report ab initio results for the structural, stability, and electronic properties for such hypothetical structures. Our results show that stable (or at least metastable) structures are possible with metallic properties. They also show that these structures can be obtained by a direct interconversion from SWNT(2,2). Large finite cubanelike oligomers, topologically related to these new tubes, were also investigated.
},
keywords = {Carbon Nanotubes, DFT, New Structures, square tubes},
pubstate = {published},
tppubtype = {article}
}
Garcez, Karl M; Moreira, Edvan; Azevedo, David L; Galvao, Douglas S
Neon atoms oscillating inside carbon and boron nitride nanotubes: a fully atomistic molecular dynamics investigation Journal Article
In: Molecular Simulation, vol. 36, no. 9, pp. 639–643, 2010.
Abstract | Links | BibTeX | Tags: Boron Nitride, Encapsulation, Molecular Dynamics, Nanotubes
@article{garcez2010neon,
title = {Neon atoms oscillating inside carbon and boron nitride nanotubes: a fully atomistic molecular dynamics investigation},
author = {Garcez, Karl M and Moreira, Edvan and Azevedo, David L and Galvao, Douglas S},
url = {http://www.tandfonline.com/doi/abs/10.1080/08927020903463926#.VLfp54rF-2o},
year = {2010},
date = {2010-01-01},
journal = {Molecular Simulation},
volume = {36},
number = {9},
pages = {639--643},
publisher = {Taylor & Francis Group},
abstract = {In the present work, based on extensive fully atomistic molecular dynamics simulations, we discuss the dynamics of neon atoms oscillating inside (5,5) single-walled carbon nanotubes (CNTs) and boron nitride nanotubes (BNNTs). Our results show that sustained high-frequency oscillatory regimes are possible for a large range of temperatures. Our results also show that the general features of the oscillations are quite similar to those observed in CNT and BNNT, in contrast with some speculations in previous works in the literature about the importance of broken symmetry and chirality exhibited by BNNTs.},
keywords = {Boron Nitride, Encapsulation, Molecular Dynamics, Nanotubes},
pubstate = {published},
tppubtype = {article}
}
Coluci, VR; dos Santos, RPB; Galvao, DS
Topologically Closed Macromolecules Made of Single Walled Carbon Nanotubes—'Super'-Fullerenes Journal Article
In: Journal of Nanoscience and Nanotechnology, vol. 10, no. 7, pp. 4378–4383, 2010.
Abstract | Links | BibTeX | Tags: Fullerenes, New Structures, Super Carbons, Super Fullerenes
@article{coluci2010topologically,
title = {Topologically Closed Macromolecules Made of Single Walled Carbon Nanotubes—'Super'-Fullerenes},
author = {Coluci, VR and dos Santos, RPB and Galvao, DS},
url = {http://www.ingentaconnect.com/content/asp/jnn/2010/00000010/00000007/art00040},
year = {2010},
date = {2010-01-01},
journal = {Journal of Nanoscience and Nanotechnology},
volume = {10},
number = {7},
pages = {4378--4383},
publisher = {American Scientific Publishers},
abstract = {We propose and theoretically investigated a new class of topologically closed macromolecules built using single walled carbon nanotubes. These macromolecules are based on the fullerene architecture. Classical molecular dynamics simulations were used to predict their stability, thermal, vibrational, and mechanical properties. These macromolecules, named 'super'-fullerenes, present high porosity, low density (∼1 g/cm3), and high surface area (≅2500 m2/g). Our results predict gas phase specific heat of about 0.4 Jg−1K−1 at room temperature and high flexibility under compressive strains. These properties make these hypothetical macromolecules good candidates for gas storage material and biomolecular sieves.},
keywords = {Fullerenes, New Structures, Super Carbons, Super Fullerenes},
pubstate = {published},
tppubtype = {article}
}
Lagos, MJ; Sato, F; Autreto, PAS; Galvao, DS; Rodrigues, V; Ugarte, D
Temperature effects on the atomic arrangement and conductance of atomic-size gold nanowires generated by mechanical stretching Journal Article
In: Nanotechnology, vol. 21, no. 48, pp. 485702, 2010.
Abstract | Links | BibTeX | Tags: DFT, Mechanical Properties, Metallic Nanowires, Quantum Transport, TEM
@article{lagos2010temperature,
title = {Temperature effects on the atomic arrangement and conductance of atomic-size gold nanowires generated by mechanical stretching},
author = {Lagos, MJ and Sato, F and Autreto, PAS and Galvao, DS and Rodrigues, V and Ugarte, D},
url = {http://iopscience.iop.org/0957-4484/21/48/485702},
year = {2010},
date = {2010-01-01},
journal = {Nanotechnology},
volume = {21},
number = {48},
pages = {485702},
publisher = {IOP Publishing},
abstract = {We have studied the changes induced by thermal effects in the structural and transport response of Au nanowires generated by mechanical elongation. We have used time-resolved atomic resolution transmission electron microscopy imaging and quantum conductance measurement using a mechanically controllable break junction. Our results showed remarkable differences in the NW evolution for experiments realized at 150 and 300 K, which modifies drastically the conductance response during elongation. Molecular dynamics and electronic transport calculations were used to consistently correlate the observed structural and conductance behavior. These results emphasize that it is essential to take into account the precise atomic arrangement of nanocontacts generated by mechanical stretching to understand electrical transport properties. Also, our study shows that much care must be taken when comparing results obtained in different experimental conditions, mainly different temperatures.
},
keywords = {DFT, Mechanical Properties, Metallic Nanowires, Quantum Transport, TEM},
pubstate = {published},
tppubtype = {article}
}
Moreira, E; Lemos, V; Galvao, DS; Azevedo, DL
$beta$-Carotene encapsulation into single-walled carbon nanotubes: a theoretical study Journal Article
In: Molecular Simulation, vol. 36, no. 13, pp. 1031–1034, 2010.
Abstract | Links | BibTeX | Tags: Beta-carotene, CNT encapsulation, Molecular Dynamics
@article{moreira2010beta,
title = {$beta$-Carotene encapsulation into single-walled carbon nanotubes: a theoretical study},
author = {Moreira, E and Lemos, V and Galvao, DS and Azevedo, DL},
url = {http://www.tandfonline.com/doi/abs/10.1080/08927022.2010.501519#.VLfmM4rF-2o},
year = {2010},
date = {2010-01-01},
journal = {Molecular Simulation},
volume = {36},
number = {13},
pages = {1031--1034},
publisher = {Taylor & Francis},
abstract = {Recently, the encapsulation of β-carotene molecules into carbon nanotubes has been achieved. In this work, we report molecular dynamics simulations and tight-binding density functional-based results for a theoretical study of the encapsulation processes. Our results show that the molecules undergo geometrical deformations when encapsulated with significant changes in their electronic structure. Based on these results, we propose a new interpretation to the changes associated with the β-carotene absorption bands experimentally observed.},
keywords = {Beta-carotene, CNT encapsulation, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Sato, F; Legoas, SB; Otero, R; Hummelink, F; Thostrup, P; Lægsgaard, E; Stensgaard, I; Besenbacher, F; Galvao, DS
Adsorption configuration effects on the surface diffusion of large organic molecules: The case of Violet Lander Journal Article
In: The Journal of chemical physics, vol. 133, no. 22, pp. 224702, 2010.
Abstract | Links | BibTeX | Tags: DFT, Diffusion, Molecular Electronics, STM, Violet Lander
@article{sato2010adsorption,
title = {Adsorption configuration effects on the surface diffusion of large organic molecules: The case of Violet Lander},
author = {Sato, F and Legoas, SB and Otero, R and Hummelink, F and Thostrup, P and Lægsgaard, E and Stensgaard, I and Besenbacher, F and Galvao, DS},
url = {http://scitation.aip.org/content/aip/journal/jcp/133/22/10.1063/1.3512623},
year = {2010},
date = {2010-01-01},
journal = {The Journal of chemical physics},
volume = {133},
number = {22},
pages = {224702},
publisher = {AIP Publishing},
abstract = {Violet Lander (C108H104) is a large organic molecule that when deposited on Cu(110) surface exhibits lock-and-key like behavior [Otero et al., Nature Mater. 3, 779 (2004)]. In this work, we report a detailed fully atomistic molecular mechanics and molecular dynamics study of this phenomenon. Our results show that it has its physical basis on the interplay of the molecular hydrogens and the Cu(110) atomic spacing, which is a direct consequence of the matching between molecule and surface dimensions. This information could be used to find new molecules capable of displaying lock-and-key behavior with new potential applications in nanotechnology.},
keywords = {DFT, Diffusion, Molecular Electronics, STM, Violet Lander},
pubstate = {published},
tppubtype = {article}
}
2009
Flores, Marcelo ZS; Autreto, Pedro AS; Legoas, Sergio B; Galvao, Douglas S
Graphene to graphane: a theoretical study Journal Article
In: Nanotechnology, vol. 20, no. 46, pp. 465704, 2009.
Abstract | Links | BibTeX | Tags: Functionalization, Graphanes, Graphene, Hydrogenation
@article{flores2009graphene,
title = {Graphene to graphane: a theoretical study},
author = {Flores, Marcelo ZS and Autreto, Pedro AS and Legoas, Sergio B and Galvao, Douglas S},
url = {http://iopscience.iop.org/0957-4484/20/46/465704},
year = {2009},
date = {2009-01-01},
journal = {Nanotechnology},
volume = {20},
number = {46},
pages = {465704},
publisher = {IOP Publishing},
abstract = {Graphane is a two-dimensional system consisting of a single layer of fully saturated (sp3 hybridization) carbon atoms. In an ideal graphane structure C–H bonds exhibit an alternating pattern (up and down with relation to the plane defined by the carbon atoms). In this work we have investigated, using ab initio and reactive molecular dynamics simulations, the role of H frustration (breaking the H atoms' up and down alternating pattern) in graphane-like structures. Our results show that a significant percentage of uncorrelated H frustrated domains are formed in the early stages of the hydrogenation process leading to membrane shrinkage and extensive membrane corrugations. These results also suggest that large domains of perfect graphane-like structures are unlikely to be formed, as H frustrated domains are always present.
},
keywords = {Functionalization, Graphanes, Graphene, Hydrogenation},
pubstate = {published},
tppubtype = {article}
}
Caetano, Ewerton WS; Freire, Valder N; Santos, Sergio G dos; Galvao, Douglas S; Sato, Fernando
Mobius and twisted graphene nanoribbons: stability, geometry and electronic properties Journal Article
In: arXiv preprint arXiv:0903.2080, 2009.
Abstract | Links | BibTeX | Tags: Graphene, Mobius, NanoRibbons, Structure
@article{caetano2009m,
title = {Mobius and twisted graphene nanoribbons: stability, geometry and electronic properties},
author = {Caetano, Ewerton WS and Freire, Valder N and Santos, Sergio G dos and Galvao, Douglas S and Sato, Fernando},
url = {http://arxiv.org/abs/0903.2080},
year = {2009},
date = {2009-01-01},
journal = {arXiv preprint arXiv:0903.2080},
abstract = {Results of classical force field geometry optimizations for twisted graphene nanoribbons with a number of twists Nt varying from 0 to 7 (the case Nt=1 corresponds to a half-twist M"obius nanoribbon) are presented in this work. Their structural stability was investigated using the Brenner reactive force field. The best classical molecular geometries were used as input for semiempirical calculations, from which the electronic properties (energy levels, HOMO, LUMO orbitals) were computed for each structure. CI wavefunctions were also calculated in the complete active space framework taking into account eigenstates from HOMO-4 to LUMO+4, as well as the oscillator strengths corresponding to the first optical transitions in the UV-VIS range. The lowest energy molecules were found less symmetric than initial configurations, and the HOMO-LUMO energy gaps are larger than the value found for the nanographene used to build them due to electronic localization effects created by the twisting. A high number of twists leads to a sharp increase of the HOMO → LUMO transition energy. We suggest that some twisted nanoribbons could form crystals stabilized by dipolar interactions.},
keywords = {Graphene, Mobius, NanoRibbons, Structure},
pubstate = {published},
tppubtype = {article}
}
Lagos, MJ; Sato, Fernando; Bettini, Jeferson; Rodrigues, Varlei; Galvao, Douglas S; Ugarte, Daniel
Observation of the smallest metal nanotube with a square cross-section Journal Article
In: Nature Nanotechnology, vol. 4, no. 3, pp. 149–152, 2009.
Abstract | Links | BibTeX | Tags: Metallic Nanowires, New Structures, Smallest nanotube, TEM, top20
@article{lagos2009observation,
title = {Observation of the smallest metal nanotube with a square cross-section},
author = {Lagos, MJ and Sato, Fernando and Bettini, Jeferson and Rodrigues, Varlei and Galvao, Douglas S and Ugarte, Daniel},
url = {http://www.nature.com/nnano/journal/v4/n3/abs/nnano.2008.414.html},
year = {2009},
date = {2009-01-01},
journal = {Nature Nanotechnology},
volume = {4},
number = {3},
pages = {149--152},
publisher = {Nature Publishing Group},
abstract = {Understanding the mechanical properties of nanoscale systems requires a range of measurement techniques and theoretical approaches to gather the relevant physical and chemical information. The arrangements of atoms in nanostructures and macroscopic matter can be different, principally due to the role of surface energy, but the interplay between atomic and electronic structure in association with applied mechanical stress can also lead to surprising differences. For example, metastable structures such as suspended chains of atoms1, 2, 3 and helical wires4, 5 have been produced by stretching metal junctions. Here, we report the spontaneous formation of the smallest possible metal nanotube with a square cross-section during the elongation of silver nanocontacts. Ab initio calculations and molecular simulations indicate that the hollow wire forms because this configuration allows the surface energy to be minimized, and also generates a soft structure capable of absorbing a huge tensile deformation.},
keywords = {Metallic Nanowires, New Structures, Smallest nanotube, TEM, top20},
pubstate = {published},
tppubtype = {article}
}
Coluci, VR; Timoteo, VS; Galvao, DS
Thermophoretically driven carbon nanotube oscillators Journal Article
In: Applied Physics Letters, vol. 95, no. 25, pp. 253103, 2009.
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, Chaos, Oscillators, Thermophoretical
@article{coluci2009thermophoretically,
title = {Thermophoretically driven carbon nanotube oscillators},
author = {Coluci, VR and Timoteo, VS and Galvao, DS},
url = {http://scitation.aip.org/content/aip/journal/apl/95/25/10.1063/1.3276546},
year = {2009},
date = {2009-01-01},
journal = {Applied Physics Letters},
volume = {95},
number = {25},
pages = {253103},
publisher = {AIP Publishing},
abstract = {The behavior of a nanodevice based upon double-walled carbon nanotubeoscillators driven by periodically applied thermal gradients (7 and 17 K/nm) is investigated by numerical calculations and classical molecular dynamics simulations. Our results indicate that thermophoresis can be effective to initiate the oscillator and that suitable heat pulses may provide an appropriate way to tune its behavior. Sustained regular oscillatory as well as chaotic motions were observed for the systems investigated in this work.},
keywords = {Carbon Nanotubes, Chaos, Oscillators, Thermophoretical},
pubstate = {published},
tppubtype = {article}
}

Perim, Eric; Galvao, Douglas S
The structure and dynamics of boron nitride nanoscrolls Journal Article
In: Nanotechnology, vol. 20, no. 33, pp. 335702, 2009.
Abstract | Links | BibTeX | Tags: Boron Nitride, Molecular Dynamics, Scrolls
@article{perim2009structure,
title = {The structure and dynamics of boron nitride nanoscrolls},
author = {Perim, Eric and Galvao, Douglas S},
url = {http://iopscience.iop.org/0957-4484/20/33/335702},
year = {2009},
date = {2009-01-01},
journal = {Nanotechnology},
volume = {20},
number = {33},
pages = {335702},
publisher = {IOP Publishing},
abstract = {Carbon nanoscrolls (CNSs) are structures formed by rolling up graphene layers into a scroll-like shape. CNNs have been experimentally produced by different groups. Boron nitride nanoscrolls (BNNSs) are similar structures using boron nitride instead of graphene layers. In this paper we report molecular mechanics and molecular dynamics results for the structural and dynamical aspects of BNNS formation. Similarly to CNS, BNNS formation is dominated by two major energy contributions, the increase in the elastic energy and the energetic gain due to van der Waals interactions of the overlapping surface of the rolled layers. The armchair scrolls are the most stable configuration while zigzag scrolls are metastable structures which can be thermally converted to armchairs. Chiral scrolls are unstable and tend to evolve into zigzag or armchair configurations depending on their initial geometries. The possible experimental routes to produce BNNSs are also addressed.
},
keywords = {Boron Nitride, Molecular Dynamics, Scrolls},
pubstate = {published},
tppubtype = {article}
}
Dos Santos, SG; Pires, MS; Lemos, V; Freire, VN; Caetano, EWS; Galvao, DS; Sato, F; Albuquerque, EL
C60-derived nanobaskets: stability, vibrational signatures, and molecular trapping Journal Article
In: Nanotechnology, vol. 20, no. 39, pp. 395701, 2009.
Abstract | Links | BibTeX | Tags: Fullerenes, Molecular Dynamics, nanobaskets, nanobowls
@article{dos2009c60,
title = {C60-derived nanobaskets: stability, vibrational signatures, and molecular trapping},
author = {Dos Santos, SG and Pires, MS and Lemos, V and Freire, VN and Caetano, EWS and Galvao, DS and Sato, F and Albuquerque, EL},
url = {http://iopscience.iop.org/0957-4484/20/39/395701},
year = {2009},
date = {2009-01-01},
journal = {Nanotechnology},
volume = {20},
number = {39},
pages = {395701},
publisher = {IOP Publishing},
abstract = {C60-derived nanobaskets, with chemical formulae (symmetry point group) C40H10 (C5v), C39H12 (C3v), C46H12 (C2v), were investigated. Molecular dynamic simulations (MDSs) indicate that the molecules preserve their bonding frame for temperatures up to 300 K (simulation time 100 ps), and maintain atomic cohesion for at least 4 ps at temperatures up to 3500 K. The infrared spectra of the C60-derived nanobaskets were simulated through density functional theory (DFT) calculations, allowing for the attribution of infrared signatures specific to each carbon nanobasket. The possibility of using C60-derived nanobaskets as molecular containers is demonstrated by performing a DFT study of their bonding to hydrogen, water, and L-alanine. The carbon nanostructures presented here show a higher bonding energy (~1.0 eV), suggesting that a family of nanostructures, Cn-derived (n = 60,70,76,80, etc) nanobaskets, could work as molecular containers, paving the way for future developments such as tunable traps for complex molecular systems.},
keywords = {Fullerenes, Molecular Dynamics, nanobaskets, nanobowls},
pubstate = {published},
tppubtype = {article}
}
Rocha, Tulio CR; Sato, Fernando; Dantas, Socrates O; Galvao, Douglas S; Zanchet, Daniela
New Insights on the Growth of Anisotropic Nanoparticles from Total Energy Calculations Journal Article
In: The Journal of Physical Chemistry C, vol. 113, no. 28, pp. 11976–11979, 2009.
Abstract | Links | BibTeX | Tags: growth, Nanoparticles, Structure
@article{rocha2009new,
title = {New Insights on the Growth of Anisotropic Nanoparticles from Total Energy Calculations},
author = {Rocha, Tulio CR and Sato, Fernando and Dantas, Socrates O and Galvao, Douglas S and Zanchet, Daniela},
url = {http://pubs.acs.org/doi/abs/10.1021/jp903794y},
year = {2009},
date = {2009-01-01},
journal = {The Journal of Physical Chemistry C},
volume = {113},
number = {28},
pages = {11976--11979},
publisher = {American Chemical Society},
abstract = {The growth mechanism of anisotropic metallic nanoparticles is still an open and polemical question. The common observation of the existence of nonspherical (not the most stable) shapes in varied experimental conditions is not fully understood. In this work, based on results from total energy calculations for different shapes and sizes of Ag nanoparticles, we provide new insights of why anisotropic structures are commonly found in different preparation conditions. We show that, assuming the presence of a particle shape distribution in the beginning of the growth process, anisotropic nanoparticles can preferentially grow over spherical ones due to the fact that the energy required to build larger anisotropic structures could be less than the one required to build isotropic structures. These results suggest that many previous works in literature shall be revisited accordingly to these new finds.},
keywords = {growth, Nanoparticles, Structure},
pubstate = {published},
tppubtype = {article}
}
Legoas, Sergio B; Autreto, Pedro AS; Flores, Marcelo ZS; Galvao, Douglas S
Graphene to graphane: the role of H frustration in lattice contraction Journal Article
In: arXiv preprint arXiv:0903.0278, 2009.
Abstract | Links | BibTeX | Tags: Functionalization, Graphane, Graphene, Hydrogenation
@article{legoas2009graphene,
title = {Graphene to graphane: the role of H frustration in lattice contraction},
author = {Legoas, Sergio B and Autreto, Pedro AS and Flores, Marcelo ZS and Galvao, Douglas S},
url = {http://arxiv.org/abs/0903.0278},
year = {2009},
date = {2009-01-01},
journal = {arXiv preprint arXiv:0903.0278},
abstract = {Graphane is a two-dimensional system consisting of a single planar layer of fully saturated (sp3 hybridization) carbon atoms with H atoms attached to them in an alternating pattern (up and down with relation to the plane defined by the carbon atoms). Stable graphane structures were theoretically predicted to exist some years ago and just experimentally realized through hydrogenation of graphene membranes. In this work we have investigated using textit{ab initio} and reactive molecular dynamics the role of H frustration (breaking the H atoms up and down alternating pattern) in graphane-like structures. Our results show that H frustration significantly contributes to lattice contraction. The dynamical aspects of converting graphene to graphane is also addressed.},
keywords = {Functionalization, Graphane, Graphene, Hydrogenation},
pubstate = {published},
tppubtype = {article}
}
Caetano, Ewerton WS; Freire, Valder N; dos Santos, Sergio G; Albuquerque, EL; Galvao, Douglas S; Sato, Fernando
Defects in Graphene-Based Twisted Nanoribbons: Structural, Electronic, and Optical Properties Journal Article
In: Langmuir, vol. 25, no. 8, pp. 4751–4759, 2009.
Abstract | Links | BibTeX | Tags: Defects, Mobius, NanoRibbons, Twisting
@article{caetano2009defects,
title = {Defects in Graphene-Based Twisted Nanoribbons: Structural, Electronic, and Optical Properties},
author = {Caetano, Ewerton WS and Freire, Valder N and dos Santos, Sergio G and Albuquerque, EL and Galvao, Douglas S and Sato, Fernando},
url = {http://pubs.acs.org/doi/abs/10.1021/la803929f},
year = {2009},
date = {2009-01-01},
journal = {Langmuir},
volume = {25},
number = {8},
pages = {4751--4759},
publisher = {ACS Publications},
abstract = {We present some computational simulations of graphene-based nanoribbons with a number of half-twists varying from 0 to 4 and two types of defects obtained by removing a single carbon atom from two different sites. Optimized geometries are found by using a mix of classical quantum semiempirical computations. According with the simulations results, the local curvature of the nanoribbons increases at the defect sites, especially for a higher number of half-twists. The HOMO−LUMO energy gap of the nanostructures has significant variation when the number of half-twists increases for the defective nanoribbons. At the quantum semiempirical level, the first optically active transitions and oscillator strengths are calculated using the full configuration interaction (CI) framework, and the optical absorption in the UV/vis range (electronic transitions) and in the infrared (vibrational transitions) are achieved. Distinct nanoribbons show unique spectral signatures in the UV/vis range, with the first absorption peaks in wavelengths ranging from the orange to the violet. Strong absorption is observed in the ultraviolet region, although differences in their infrared spectra are hardly discernible.},
keywords = {Defects, Mobius, NanoRibbons, Twisting},
pubstate = {published},
tppubtype = {article}
}
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ

