
Borges, Daiane Damasceno; Galvao, Douglas S.
Schwarzites for Natural Gas Storage: A Grand-Canonical Monte Carlo Study Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 115-120, 2018.
@article{Borges2018d,
title = {Schwarzites for Natural Gas Storage: A Grand-Canonical Monte Carlo Study },
author = {Daiane Damasceno Borges and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/schwarzites-for-natural-gas-storage-a-grandcanonical-monte-carlo-study/2DF8D601AF8EF04BBAC5CCCBEFA8339E},
doi = {https://doi.org/10.1557/adv.2018.190},
year = {2018},
date = {2018-02-13},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = {115-120},
abstract = {he 3D porous carbon-based structures called Schwarzites have been recently a subject of renewed interest due to the possibility of being synthesized in the near future. These structures exhibit negatively curvature topologies with tuneable porous sizes and shapes, which make them natural candidates for applications such as CO2 capture, gas storage and separation. Nevertheless, the adsorption properties of these materials have not been fully investigated. Following this motivation, we have carried out Grand-Canonical Monte Carlo simulations to study the adsorption of small molecules such as CO2, CO, CH4, N2 and H2, in a series of Schwarzites structures. Here, we present our preliminary results on natural gas adsorptive capacity in association with analyses of the guest-host interaction strengths. Our results show that Schwarzites P7par, P8bal and IWPg are the most promising structures with very high CO2 and CH4 adsorption capacity and low saturation pressure (<1bar) at ambient temperature. The P688 is interesting for H2 storage due to its exceptional high H2 adsorption enthalpy value of -19kJ/mol.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Borges, Daiane Damasceno; Woellner, Cristiano F.; Autreto, Pedro A. S.; Galvao, Douglas S.
Water/alcohol separation via layered oxide graphene membranes Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 109-114, 2018.
@article{Borges2018d,
title = {Water/alcohol separation via layered oxide graphene membranes},
author = {Daiane Damasceno Borges and Cristiano F. Woellner and Pedro A. S. Autreto and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/wateralcohol-separation-in-graphene-oxide-membranes-insights-from-molecular-dynamics-and-monte-carlo-simulations/C61C66FF48D35EB2DB3408ACCE96C41A},
doi = { https://doi.org/10.1557/adv.2018.192},
year = {2018},
date = {2018-02-13},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = {109-114},
abstract = {Graphene-based membranes have been investigated as promising candidates for water filtration and gas separation applications. Experimental evidences have shown that graphene oxide can be impermeable to liquids, vapors and gases, while allowing a fast permeation of water molecules. This phenomenon has been attributed to the formation of a network of nano capillaries that allow nearly frictionless water flow while blocking other molecules by steric hindrance effects. It is supposed that water molecules are transported through the percolated two-dimensional channels formed between graphene-based sheets. Although these channels allow fast water permeation in such materials, the flow rates are strongly dependent on how the membranes are fabricated. Also, some fundamental issues regarding the nanoscale mechanisms of water permeation are still not fully understood and their interpretation remains controversial. In this work, we have investigated the dynamics of water permeation through pristine graphene and graphene oxide model membranes that have strong impact on water/alcohol separation. We have carried out fully atomistic classical molecular dynamics simulations of systems composed of multiple layered graphene-based sheets into contact with a pure water reservoir under controlled thermodynamics conditions (e. g., by varying temperature and pressure values). We have systematically analysed how the transport dynamics of the confined nanofluids depend on the interlayer distances and the role of the oxide functional groups. Our results show the water flux is much more effective for graphene than for graphene oxide membranes. These results can be attributed to the H-bonds formation between oxide functional groups and water, which traps the water molecules and precludes ultrafast water transport through the nanochannels.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
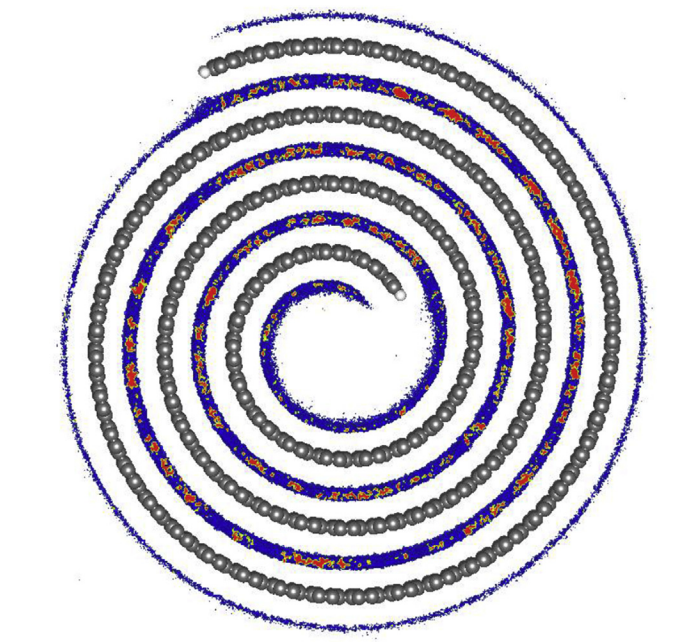
Shadmi, Nitzan; Kremen, Anna; Frenkel, Yiftach; Lapin, Zachary J.; Machado, Leonardo D.; Legoas, Sergio B.; Bitton, Ora; Rechav, Katya; Popovitz-Biro, Ronit; Galvão, Douglas S.; Jorio, Ado; Novotny, Lukas; Kalisky, Beena; Joselevich, Ernesto
Defect-Free Carbon Nanotube Coils Online
2018, (reprint Nano Letters v16, 2152 (2016)).
@online{Shadmi2018,
title = {Defect-Free Carbon Nanotube Coils },
author = {Nitzan Shadmi and Anna Kremen and Yiftach Frenkel and Zachary J. Lapin and Leonardo D. Machado and Sergio B. Legoas and Ora Bitton and Katya Rechav and Ronit Popovitz-Biro and Douglas S. Galvão and Ado Jorio and Lukas Novotny and Beena Kalisky and Ernesto Joselevich},
url = {https://arxiv.org/abs/1802.03715},
year = {2018},
date = {2018-02-13},
abstract = {Carbon nanotubes are promising building blocks for various nanoelectronic components. A
highly desirable geometry for such applications is a coil. However, coiled nanotube structures
reported so far were inherently defective or had no free ends accessible for contacting. Here we
demonstrate the spontaneous self-coiling of single-wall carbon nanotubes into defect-free coils
of up to more than 70 turns with identical diameter and chirality, and free ends. We characterize
the structure, formation mechanism and electrical properties of these coils by different
microscopies, molecular dynamics simulations, Raman spectroscopy, and electrical and magnetic
measurements. The coils are highly conductive, as expected for defect-free carbon nanotubes,
but adjacent nanotube segments in the coil are more highly coupled than in regular bundles of
single-wall carbon nanotubes, owing to their perfect crystal momentum matching, which enables
tunneling between the turns. Although this behavior does not yet enable the performance of these
nanotube coils as inductive devices, it does point a clear path for their realization. Hence, this
study represents a major step toward the production of many different nanotube coil devices,
including inductors, electromagnets, transformers and dynamos.},
note = {reprint Nano Letters v16, 2152 (2016)},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
highly desirable geometry for such applications is a coil. However, coiled nanotube structures
reported so far were inherently defective or had no free ends accessible for contacting. Here we
demonstrate the spontaneous self-coiling of single-wall carbon nanotubes into defect-free coils
of up to more than 70 turns with identical diameter and chirality, and free ends. We characterize
the structure, formation mechanism and electrical properties of these coils by different
microscopies, molecular dynamics simulations, Raman spectroscopy, and electrical and magnetic
measurements. The coils are highly conductive, as expected for defect-free carbon nanotubes,
but adjacent nanotube segments in the coil are more highly coupled than in regular bundles of
single-wall carbon nanotubes, owing to their perfect crystal momentum matching, which enables
tunneling between the turns. Although this behavior does not yet enable the performance of these
nanotube coils as inductive devices, it does point a clear path for their realization. Hence, this
study represents a major step toward the production of many different nanotube coil devices,
including inductors, electromagnets, transformers and dynamos.
Oliveira, Eliezer Fernando; Autreto, Pedro Alves da Silva; Galvao, Douglas Soares
On hardening silver nanocubes by high velocity impacts: a fully atomistic molecular dynamics investigation Journal Article
In: Journal of Materials Science, vol. 53, no. 10, pp. 7486–7492, 2018.
@article{Oliveira2018,
title = {On hardening silver nanocubes by high velocity impacts: a fully atomistic molecular dynamics investigation},
author = {Oliveira, Eliezer Fernando and Autreto, Pedro Alves da Silva and Galvao, Douglas Soares},
url = {https://link.springer.com/article/10.1007/s10853-018-2104-z},
doi = {10.1007/s10853-018-2104-z},
year = {2018},
date = {2018-02-09},
journal = {Journal of Materials Science},
volume = {53},
number = {10},
pages = {7486–7492},
abstract = {Gradient nanograins (GNG) creation in metals has been a promising approach to obtain ultra-strong materials. Recently, R. Thevamaran et al. (Science 354:312 in 2016) proposed a single-step method based on high-velocity impacts of silver nanocubes (SNC) to produce almost perfect GNG. However, after certain time, these grains spontaneously coalesce, which compromises the induced hardening and other mechanical properties. To better understand these processes, a detailed investigation at the atomic scale of the deformation/hardening mechanisms are needed, which is one of the objectives of the present work. We carried out fully atomistic molecular dynamics (MD) simulations of silver nanocubes at high impact velocity values using realistic structural models. Our MD results suggest that besides the GNG mechanisms, the observed SNC hardening could be also the result of the existence of polycrystalline arrangements formed by HCP domains encapsulated by FCC ones in the smashed SNC. This can be a new way to design ultra-strong materials, even in the absence of GNG domains.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
de Sousa, J. M.; Aguiar, A. L.; Girao, E. C.; Fonseca, Alexandre F.; Filho, A. G. Souza; Galvao, Douglas S.
Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 97-102, 2018.
@article{deSousa2018b,
title = {Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study},
author = {J. M. de Sousa and A. L. Aguiar and E. C. Girao and Alexandre F. Fonseca and A. G. Souza Filho and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-pentagraphenebased-nanotubes-a-molecular-dynamics-study/289AB70DADF20059BB8FCC9EF07B97AB},
doi = { https://doi.org/10.1557/adv.2018.160},
year = {2018},
date = {2018-02-06},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = {97-102},
abstract = {The study of the mechanical properties of nanostructured systems has gained importance in theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of the strongest nanomaterials found in nature, with Young’s Modulus (EY) in the order 1.25 TPa. One interesting question is about the possibility of generating new nanostructures with 1D symmetry and with similar and/or superior CNT properties. In this work, we present a study on the dynamical, structural, mechanical properties, fracture patterns and EY values for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized states) in the same form that CNTs are formed from rolling up graphene membranes. We carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We have considered zigzag-like and armchair-like PGNTs of different diameters. Our results show that PGNTs present EY ∼ 800 GPa with distinct elastic behavior in relation to CNTs, mainly associated with mechanical failure, chirality dependent fracture patterns and extensive structural reconstructions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Cristiano F Woellner Daiane Damasceno Borges, Pedro AS Autreto
Insights on the mechanism of water-alcohol separation in multilayer graphene oxide membranes: entropic versus enthalpic factors Journal Article
In: Carbon, vol. 127, pp. 280-286, 2018.
@article{Borges2018,
title = {Insights on the mechanism of water-alcohol separation in multilayer graphene oxide membranes: entropic versus enthalpic factors},
author = {Daiane Damasceno Borges, Cristiano F Woellner, Pedro AS Autreto, Douglas S Galvao},
url = {https://www.sciencedirect.com/science/article/pii/S000862231731134X},
doi = {https://doi.org/10.1016/j.carbon.2017.11.020},
year = {2018},
date = {2018-02-01},
journal = {Carbon},
volume = {127},
pages = {280-286},
abstract = {xperimental evidence has shown that graphene oxide (GO) can be impermeable to liquids, vapors and gases, while it allows a fast permeation of water molecules. Theoretical studies to understand the filtration mechanisms come mostly from water desalination, while very few works have been dedicated to alcohol dehydration. In this work, we have investigated the molecular level mechanism underlying the alcohol/water separation inside GO membranes. A series of Molecular Dynamics and Grand-Canonical Monte Carlo simulations were carried out to probe the ethanol/water and methanol/water separation through GO membranes composed of multiple layered graphene-based films with different interlayer distance values and number of oxygen-containing functional groups. Our results show that the size exclusion and membrane affinities are not sufficient to explain the selectivity. Besides that, the favorable water molecular arrangement inside GO 2D-channels forming a robust H-bond network and the fast water permeation are crucial for an effective separation mechanism. In other words, the separation phenomenon is not only governed by membrane affinities (enthalpic mechanisms) but mainly by the geometry and size factors (entropic mechanisms). Our findings are consistent with the available experimental data and contribute to clarify important aspects of the separation behavior of confined alcohol/water in GO membranes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Fonseca, Alexandre F.; Galvao, Douglas S.
Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation Journal Article
In: MRS Advances, vol. 3, no. 8-9, pp. 460-465, 2018.
@article{Fonseca2018,
title = {Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation},
author = {Alexandre F. Fonseca and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/selfdriven-graphene-tearing-and-peeling-a-fully-atomistic-molecular-dynamics-investigation/BFC76FC4479AA617E16FA6AC7AB4D487},
doi = {https://doi.org/10.1557/adv.2018.120},
year = {2018},
date = {2018-01-30},
journal = {MRS Advances},
volume = {3},
number = {8-9},
pages = {460-465},
abstract = {In spite of years of intense research, graphene continues to produce surprising results. Recently, it was experimentally observed that under certain conditions graphene can self-drive its tearing and peeling from substrates. This process can generate long, micrometer sized, folded nanoribbons without the action of any external forces. Also, during this cracking-like propagation process, the width of the graphene folded ribbon continuously decreases and the process only stops when the width reaches about few hundreds nanometers in size. It is believed that interplay between the strain energy of folded regions, breaking of carbon-carbon covalent bonds, and adhesion of graphene-graphene and graphene-substrate are the most fundamental features of this process, although the detailed mechanisms at atomic scale remain unclear. In order to gain further insights on these processes we carried out fully atomistic reactive molecular dynamics simulations using the AIREBO potential as available in the LAMMPS computational package. Although the reported tearing/peeling experimental observations were only to micrometer sized structures, our results showed that they could also occur at nanometer scale. Our preliminary results suggest that the graphene tearing/peeling process originates from thermal energy fluctuations that results in broken bonds, followed by strain release that creates a local elastic wave that can either reinforce the process, similar to a whip cracking propagation, or undermine it by producing carbon dangling bonds that evolve to the formation of bonds between the two layers of graphene. As the process continues in time and the folded graphene decreases in width, the carbon-carbon bonds at the ribbon edge and interlayer bonds get less stressed, thermal fluctuations become unable to break them and the process stops.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Woellner, Cristiano F.; Botari, Tiago; Perim, Eric; Galvao, Douglas S.
Mechanical Properties of Schwarzites - A Fully Atomistic Reactive Molecular Dynamics Investigation Journal Article
In: MRS Advances, pp. 1-6, 2018.
@article{Woellner2018b,
title = {Mechanical Properties of Schwarzites - A Fully Atomistic Reactive Molecular Dynamics Investigation},
author = {Cristiano F. Woellner and Tiago Botari and Eric Perim and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-schwarzites-a-fully-atomistic-reactive-molecular-dynamics-investigation/012AF477491A46541A052C944E4E4834},
doi = { https://doi.org/10.1557/adv.2018.124},
year = {2018},
date = {2018-01-29},
journal = {MRS Advances},
pages = {1-6},
abstract = {Schwarzites are crystalline, 3D porous structures with a stable negative curvature formed of sp2-hybridized carbon atoms. These structures present topologies with tunable porous size and shape and unusual mechanical properties. In this work, we have investigated the mechanical behavior under compressive strain and energy absorption of four different Schwarzites. We considered two Schwarzites families, the so-called Gyroid and Primitive and two structures from each family. We carried out reactive molecular dynamics simulations, using the ReaxFF force field as available in the LAMMPS code. Our results also show they exhibit remarkable resilience under mechanical compression. They can be reduced to half of their original size before structural failure (fracture) occurs.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Woellner, Cristiano F.; Owuor, Peter S.; Li, Tong; Vinod, Soumya; Ozden, Sehmus; Kosolwattana, Suppanat; Bhowmick, Sanjit; Duy, Luong X.; Salvatierra, Rodrigo V.; Wei, Bingqing; Asif, Syed A. S.; Tour, James M.; Vajtai, Robert; Lou, Jun; Galvão, Douglas S.; Tiwary, Chandra S.; Ajayan, Pulickel. M.
Mechanical Properties of Ultralow Density Graphene Oxide/Polydimethylsiloxane Foams Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 61-66, 2018.
@article{Woellner2018c,
title = {Mechanical Properties of Ultralow Density Graphene Oxide/Polydimethylsiloxane Foams},
author = {Cristiano F. Woellner and Peter S. Owuor and Tong Li and Soumya Vinod and Sehmus Ozden and Suppanat Kosolwattana and Sanjit Bhowmick and Luong X. Duy and Rodrigo V. Salvatierra and Bingqing Wei and Syed A. S. Asif and James M. Tour and Robert Vajtai and Jun Lou and Douglas S. Galvão and Chandra S. Tiwary and Pulickel. M. Ajayan},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-ultralow-density-graphene-oxidepolydimethylsiloxane-foams/BC2DC24B3DB5714759FC1EDC71BD9D05},
doi = {DOI: 10.1557/adv.2018. 49},
year = {2018},
date = {2018-01-18},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = { 61-66},
abstract = {Low-density, highly porous graphene/graphene oxide (GO) based-foams have shown high performance in energy absorption applications, even under high compressive deformations. In general, foams are very effective as energy dissipative materials and have been widely used in many areas such as automotive, aerospace and biomedical industries. In the case of graphene-based foams, the good mechanical properties are mainly attributed to the intrinsic graphene and/or GO electronic and mechanical properties. Despite the attractive physical properties of graphene/GO based-foams, their structural and thermal stabilities are still a problem for some applications. For instance, they are easily degraded when placed in flowing solutions, either by the collapsing of their layers or just by structural disintegration into small pieces. Recently, a new and scalable synthetic approach to produce low-density 3D macroscopic GO structure interconnected with polydimethylsiloxane (PDMS) polymeric chains (pGO) was proposed. A controlled amount of PDMS is infused into the freeze-dried foam resulting into a very rigid structure with improved mechanical properties, such as tensile plasticity and toughness. The PDMS wets the graphene oxide sheets and acts like a glue bonding PDMS and GO sheets. In order to obtain further insights on mechanisms behind the enhanced mechanical pGO response we carried out fully atomistic molecular dynamics (MD) simulations. Based on MD results, we build up a structural model that can explain the experimentally observed mechanical behavior.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Azevedo, David L.; Bizao, Rafael A.; Galvao, Douglas S.
Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets Online
2018, (preprint arXiv:1801.05346).
@online{Azevedo2018b,
title = {Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets},
author = {David L. Azevedo and Rafael A. Bizao and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.05346},
year = {2018},
date = {2018-01-18},
abstract = {The superior mechanical properties and low density of carbon nanostructures make them promising ballistic protection materials, stimulating investigations on their high-strain-rate behavior. Recent experiments and simulations revealed graphene possesses exceptional energy absorption properties. In this work, we analyzed through fully atomistic molecular dynamics simulations the ballistic performance of a carbon-based material recently proposed named penta-graphene. Our results show that the fracture pattern is more spherical (no petals formation like observed for graphene). The estimated penetration energy for pentagraphene structures considered here was of 37.69 MJ/Kg, far superior to graphene (29.8 MJ/Kg) under same conditions. These preliminary results are suggestive that pentagraphene could be an excellent material for ballistic applications.},
note = {preprint arXiv:1801.05346},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Woellner, Cristiano F.; Botari, Tiago; Perim, Eric; Galvao, Douglas S.
Mechanical Properties of Schwarzites - A Fully Atomistic Reactive Molecular Dynamics Investigation Online
2018, (preprint arXiv:1801.05639).
@online{Woellner2018d,
title = {Mechanical Properties of Schwarzites - A Fully Atomistic Reactive Molecular Dynamics Investigation},
author = {Cristiano F. Woellner and Tiago Botari and Eric Perim and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.05639},
year = {2018},
date = {2018-01-18},
abstract = {Schwarzites are crystalline, 3D porous structures with stable negative curvature formed of sp2-hybridized carbon atoms. These structures present topologies with tunable porous size and shape and unusual mechanical properties. In this work, we have investigated the mechanical behavior under compressive strains and energy absorption of four different Schwarzites, through reactive molecular dynamics simulations, using the ReaxFF force field as available in the LAMMPS code. We considered two Schwarzites families, the so-called Gyroid and Primitive and two structures from each family. Our results also show they exhibit remarkable resilience under mechanical compression. They can be reduced to half of their original size before structural failure (fracture) occurs.},
note = {preprint arXiv:1801.05639},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Jaques, Y. M.; Manimunda, P.; Nakanishi, Y.; Susarla, S.; Woellner, C. F.; Bhowmick, S.; Asif, S. A. S.; Galvao, D. S.; C. S. Tiwary,; Ajayan, P. M.
Differences in the Mechanical Properties of Monolayer and Multilayer WSe2/MoSe2 Online
2018, (preprint arXiv:1801.05641).
@online{Jaques2018b,
title = {Differences in the Mechanical Properties of Monolayer and Multilayer WSe2/MoSe2},
author = {Y. M. Jaques and P. Manimunda and Y. Nakanishi and S. Susarla and C. F. Woellner and S. Bhowmick and S. A. S. Asif and D. S. Galvao and C. S. Tiwary, and P. M. Ajayan},
url = {https://arxiv.org/abs/1801.05641},
year = {2018},
date = {2018-01-18},
abstract = {Transition metal dichalcogenides are 2D structures with remarkable electronic, chemical, optical and mechanical properties. Monolayer and crystal properties of these structures have been extensively investigated, but a detailed understanding of the properties of their few-layer structures are still missing. In this work we investigated the mechanical differences between monolayer and multilayer WSe2 and MoSe2, through fully atomistic molecular dynamics simulations (MD). It was observed that single layer WSe2/MoSe2 deposited on silicon substrates have larger friction coefficients than 2, 3 and 4 layered structures. For all considered cases it is always easier to peel off and/or to fracture MoSe2 structures. These results suggest that the interactions between first layer and substrate are stronger than interlayer interactions themselves. Similar findings have been reported for other nanomaterials and it has been speculated whether this is a universal-like behavior for 2D layered materials. We have also analyzed fracture patterns. Our results show that fracture is chirality dependent with crack propagation preferentially perpendicular to W(Mo)-Se bonds and faster for zig-zag-like defects.},
note = {preprint arXiv:1801.05641},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Fonseca, Alexandre F.; Galvao, Douglas S.
Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation Online
2018, (preprint arXiv:1801.05354).
@online{Fonseca2018b,
title = {Self-Driven Graphene Tearing and Peeling: A Fully Atomistic Molecular Dynamics Investigation},
author = {Alexandre F. Fonseca and Douglas S. Galvao
},
url = {https://arxiv.org/abs/1801.05354},
year = {2018},
date = {2018-01-17},
abstract = {In spite of years of intense research, graphene continues to produce surprising results. Recently, it was experimentally observed that under certain conditions graphene can self-drive its tearing and peeling from substrates. This process can generate long, micrometer sized, folded nanoribbons without the action of any external forces. Also, during this cracking-like propagation process, the width of the graphene folded ribbon continuously decreases and the process only stops when the width reaches about few hundreds nanometers in size. It is believed that interplay between the strain energy of folded regions, breaking of carbon-carbon covalent bonds, and adhesion of graphene-graphene and graphene-substrate are the most fundamental features of this process, although the detailed mechanisms at atomic scale remain unclear. In order to gain further insights on these processes we carried out fully atomistic reactive molecular dynamics simulations using the AIREBO potential as available in the LAMMPS computational package. Although the reported tearing/peeling experimental observations were only to micrometer sized structures, our results showed that they could also occur at nanometer scale. Our preliminary results suggest that the graphene tearing/peeling process originates from thermal energy fluctuations that results in broken bonds, followed by strain release that creates a local elastic wave that can either reinforce the process, similar to a whip cracking propagation, or undermine it by producing carbon dangling bonds that evolve to the formation of bonds between the two layers of graphene. As the process continues in time and the folded graphene decreases in width, the carbon-carbon bonds at the ribbon edge and interlayer bonds get less stressed, thermal fluctuations become unable to break them and the process stops.},
note = {preprint arXiv:1801.05354},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
de Sousa, J. M.; Aguiar, A. L.; Girao, E. C.; Fonseca, Alexandre F.; Filho, A. G. Souza; Galvao, Douglas S.
Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation Journal Article
In: MRS Advances, vol. 3, no. 1-2, pp. 67-72, 2018.
@article{deSousa2018c,
title = {Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation},
author = {J. M. de Sousa and A. L. Aguiar and E. C. Girao and Alexandre F. Fonseca and A. G. Souza Filho and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/mechanical-properties-of-phagraphene-membranes-a-fully-atomistic-molecular-dynamics-investigation/3ADC3F3B0052AB6632E8681404948E7B},
doi = {DOI: 10.1557/adv.2018. 54},
year = {2018},
date = {2018-01-15},
journal = {MRS Advances},
volume = {3},
number = {1-2},
pages = {67-72},
abstract = {Recently, a new 2D carbon allotrope structure, named phagraphene (PG), was proposed. PG has a densely array of penta-hexa-hepta-graphene carbon rings. PG was shown to present low and anisotropic thermal conductivity and it is believed that this anisotropy should be also reflected in its mechanical properties. Although PG mechanical properties have been investigated, a detailed and comprehensive study is still lacking. In the present work we have carried out fully atomistic reactive molecular dynamics simulations using the ReaxFF force field, to investigate the mechanical properties and fracture patterns of PG membranes. The Young's modulus values of the PG membranes were estimated from the stress-strain curves. Our results show that these curves present three distinct regimes: one regime where ripples dominate the structure and mechanical properties of the PG membranes; an elastic regime where the membranes exhibit fully planar configurations; and finally am inelastic regime where permanent deformations happened to the PG membrane up to the mechanical failure or fracture.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
de Sousa, J. M.; Aguiar, A. L.; Girao, E. C.; Fonseca, Alexandre F.; Filho, A. G. Sousa; Galvao, Douglas S.
Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study Online
2018, (preprint arXiv:1801.04269).
@online{deSousa2018d,
title = {Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study},
author = {J. M. de Sousa and A. L. Aguiar and E. C. Girao and Alexandre F. Fonseca and A. G. Sousa Filho and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.04269},
year = {2018},
date = {2018-01-15},
abstract = {The study of the mechanical properties of nanostructured systems has gained importance in
theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of
the strongest nanomaterials found in nature, with Young's Modulus (YM) in the order 1.25
TPa. One interesting question is about the possibility of generating new nanostructures with
1D symmetry and with similar and/or superior CNT properties. In this work, we present a
study on the dynamical, structural, mechanical properties, fracture patterns and YM values
for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These
tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional
structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized
states) in the same form that CNTs are formed from rolling up graphene membranes. We
carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We
have considered zigzag-like and armchair-like PGNTs of different diameters. Our results
show that PGNTs present YM ~ 800 GPa with distinct elastic behavior in relation to CNTs,
mainly associated with mechanical failure, chirality dependent fracture patterns and extensive
structural reconstructions},
note = {preprint arXiv:1801.04269},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of
the strongest nanomaterials found in nature, with Young's Modulus (YM) in the order 1.25
TPa. One interesting question is about the possibility of generating new nanostructures with
1D symmetry and with similar and/or superior CNT properties. In this work, we present a
study on the dynamical, structural, mechanical properties, fracture patterns and YM values
for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These
tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional
structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized
states) in the same form that CNTs are formed from rolling up graphene membranes. We
carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We
have considered zigzag-like and armchair-like PGNTs of different diameters. Our results
show that PGNTs present YM ~ 800 GPa with distinct elastic behavior in relation to CNTs,
mainly associated with mechanical failure, chirality dependent fracture patterns and extensive
structural reconstructions
Oliveira, Eliezer Fernando; Santos, Ricardo Paupitz; da Silva Autreto, Pedro Alves; Stanislav Moshkalev,; Galvao, Douglas Soares
Improving Graphene-metal Contacts: Thermal Induced Polishing Online
2018, (preprint ArXiv:1801.04785).
@online{Oliveira2018d,
title = {Improving Graphene-metal Contacts: Thermal Induced Polishing},
author = {Eliezer Fernando Oliveira and Ricardo Paupitz Santos and Pedro Alves da Silva Autreto and Stanislav Moshkalev, and Douglas Soares Galvao},
url = {https://arxiv.org/abs/1801.04785},
year = {2018},
date = {2018-01-15},
abstract = {Graphene is a very promising material for nanoelectronics applications due to its unique and remarkable electronic and thermal properties. However, when deposited on metallic electrodes the overall thermal conductivity is significantly decreased. This phenomenon has been attributed to the mismatch between the interfaces and contact thermal resistance. Experimentally, one way to improve the graphene/metal contact is thorough high-temperature annealing, but the detailed mechanisms behind these processes remain unclear. In order to address these questions, we carried out fully atomistic reactive molecular dynamics simulations using the ReaxFF force field to investigate the interactions between multi-layer graphene and metallic electrodes (nickel) under (thermal) annealing. Our results show that the annealing induces an upward-downward movement of the graphene layers, causing a pile- driver-like effect over the metallic surface. This graphene induced movements cause a planarization (thermal polishing-like effect) of the metallic surface, which results in the increase of the effective graphene/metal contact area. This can also explain the experimentally observed improvements of the thermal and electric conductivities.},
note = {preprint ArXiv:1801.04785},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Owuor, Peter; Chaudhary, Varun; Woellner, Cristiano F; Ramanujan, R V; Stender, Anthony S; Soto, Matias; Ozden, Sehmus; Barrera, Enrique; Vajtai, Robert; Galvao, Douglas; Lou, Jun; Sharma, V; Ajayan, Pulickel M
High Stiffness Polymer Composite with Tunable Transparency Journal Article
In: Materials Today, vol. 21, no. 5, pp. 475-482, 2018.
@article{Owuor2018,
title = {High Stiffness Polymer Composite with Tunable Transparency},
author = {Peter Owuor and Varun Chaudhary and Cristiano F Woellner and R V Ramanujan and Anthony S Stender and Matias Soto and Sehmus Ozden and Enrique Barrera and Robert Vajtai and Douglas Galvao and Jun Lou and V Sharma and Pulickel M Ajayan
},
url = {https://www.sciencedirect.com/science/article/pii/S1369702117306867},
doi = {10.1016/j.mattod.2017.12.004},
year = {2018},
date = {2018-01-12},
journal = {Materials Today},
volume = {21},
number = {5},
pages = {475-482},
abstract = {Biological materials are multifunctional performing more than one function in a perfect synergy. These materials are built from fairly simple and limited components at ambient conditions. Such judicious designs have proven elusive for synthetic materials. Here, we demonstrate a multifunctional phase change (pc) composite from simple building blocks, which exhibits high stiffness and optical transmittance control. We show an increase of more than one order of magnitude in stiffness when we embed paraffin wax spheres into an elastomer matrix, polydimethylsiloxane (PDMS) in a dynamic compression test. High stiffness is mainly influenced by presence of microcrystals within the wax. We further show fast temperature-controlled optical switching of the composite for an unlimited number of cycles without any noticeable mechanical degradation. Through experimental and finite element method, we show high energy absorption capability of pc-composite. Based on these properties, the pc- composite could be used as an effective coating on glasses for cars and windows. This simple approach to multi-functionality is exciting and could pave way for designs of other multifunctional materials at the macro-scale.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
de Sousa, Jose M.; Aguiar, Acrisio L.; Girao, Eduardo C.; Fonseca, Alexandre F.; Antonio G. Sousa Filho,; Galvao, Douglas S.
Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation Online
2018, (preprint arXiv:1801.04292).
@online{deSousa2018e,
title = {Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Jose M. de Sousa and Acrisio L. Aguiar and Eduardo C. Girao and Alexandre F. Fonseca and Antonio G. Sousa Filho, and Douglas S. Galvao
},
url = {https://arxiv.org/abs/1801.04292},
year = {2018},
date = {2018-01-12},
abstract = {Recently, a new 2D carbon allotrope structure, named phagraphene (PG), was proposed. PG has a densely array of penta-hexa-hepta-graphene carbon rings. PG was shown to present low and anisotropic thermal conductivity and it is believed that this anisotropy should be also reflected in its mechanical properties. Although PG mechanical properties have been investigated, a detailed and comprehensive study is still lacking. In the present work we have carried out fully atomistic reactive molecular dynamics simulations using the ReaxFF force field, to investigate the mechanical properties and fracture patterns of PG membranes. The Young's modulus values of the PG membranes were estimated from the stress-strain curves. Our results show that these curves present three distinct regimes: one regime where ripples dominate the structure and mechanical properties of the PG membranes; an elastic regime where the membranes exhibit fully planar configurations; and finally a plastic regime where permanent deformations happened to the PG membrane up to the mechanical failure or fracture.},
note = {preprint arXiv:1801.04292},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
de Sousa, Jose M.; Aguiar, Acrisio L.; Girao, Eduardo C.; Fonseca, Alexandre F.; Antonio G. Souza Filho,; Galvao, Douglas S.
Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study Online
2018, (preprint arXiv:1801.04269).
@online{deSousa2018f,
title = {Mechanical Properties of Pentagraphene-based Nanotubes: A Molecular Dynamics Study},
author = {Jose M. de Sousa and Acrisio L. Aguiar and Eduardo C. Girao and Alexandre F. Fonseca and Antonio G. Souza Filho, and Douglas S. Galvao},
url = {https://arxiv.org/abs/1801.04269},
year = {2018},
date = {2018-01-12},
abstract = {The study of the mechanical properties of nanostructured systems has gained importance in theoretical and experimental research in recent years. Carbon nanotubes (CNTs) are one of the strongest nanomaterials found in nature, with Young's Modulus (YM) in the order 1.25 TPa. One interesting question is about the possibility of generating new nanostructures with 1D symmetry and with similar and/or superior CNT properties. In this work, we present a study on the dynamical, structural, mechanical properties, fracture patterns and YM values for one class of these structures, the so-called pentagraphene nanotubes (PGNTs). These tubes are formed rolling up pentagraphene membranes (which are quasi-bidimensional structures formed by densely compacted pentagons of carbon atoms in sp3 and sp2 hybridized states) in the same form that CNTs are formed from rolling up graphene membranes. We carried out fully atomistic molecular dynamics simulations using the ReaxFF force field. We have considered zigzag-like and armchair-like PGNTs of different diameters. Our results show that PGNTs present YM ~ 800 GPa with distinct elastic behavior in relation to CNTs, mainly associated with mechanical failure, chirality dependent fracture patterns and extensive structural reconstructions.},
note = {preprint arXiv:1801.04269},
keywords = {},
pubstate = {published},
tppubtype = {online}
}
Azevedo, David L.; Bizao, Rafael A.; Galvao, Douglas S.
Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets Journal Article
In: MRS Advances, vol. 3, no. 8-9, pp. 431-435, 2018.
@article{Azevedo2018,
title = {Molecular Dynamics Simulations of Ballistic Penetration of Pentagraphene Sheets},
author = {David L. Azevedo and Rafael A. Bizao and Douglas S. Galvao},
url = {https://www.cambridge.org/core/journals/mrs-advances/article/molecular-dynamics-simulations-of-ballistic-penetration-of-pentagraphene-sheets/8759C0815840EDE83896EF4A17278228},
doi = {https://doi.org/10.1557/adv.2018.61},
year = {2018},
date = {2018-01-06},
journal = {MRS Advances},
volume = {3},
number = {8-9},
pages = {431-435},
abstract = {The search for new materials with low density and superior mechanical properties is a very intense and stimulating investigation area. These new materials could provide potential application for ballistic protection. Recent experiments and simulations revealed graphene possesses exceptional energy absorption properties. In this work, we analysed through fully atomistic molecular dynamics simulations the ballistic performance of a carbon-based material recently proposed named penta-graphene. Our results show that the fracture pattern is more spherical (no petals formation like observed for graphene). The estimated penetration energy for single-layer penta-graphene structures obtained here was d_1penta∼37.7 MJ/kg, and is comparable with recently results obtained for graphene: d_(1graphene)∼29.0 MJ/kg and d_(1graphene)∼40.8 MJ/kg under similar conditions. These preliminary results are suggestive that penta-graphene could be an excellent material for ballistic applications.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2016

Anna Kremen Nitzan Shadmi, Yiftach Frenkel; Joselevich, Ernesto
Defect-Free Carbon Nanotube Coils Journal Article
In: Nano Letters, vol. 16, no. 4, pp. 2152–2158, 2016.
Abstract | Links | BibTeX | Tags: CNT, Coils, Molecular Dynamics, Synthesis, TEM
@article{Shadmi2016,
title = {Defect-Free Carbon Nanotube Coils},
author = {Nitzan Shadmi, Anna Kremen, Yiftach Frenkel, Zachary J. Lapin, Leonardo D. Machado, Sergio B. Legoas, Ora Bitton, Katya Rechav, Ronit Popovitz-Biro, Douglas S. Galvão, Ado Jorio, Lukas Novotny, Beena Kalisky, and Ernesto Joselevich},
url = {http://pubs.acs.org/doi/abs/10.1021/acs.nanolett.5b03417},
doi = {10.1021/acs.nanolett.5b03417},
year = {2016},
date = {2016-04-01},
journal = {Nano Letters},
volume = {16},
number = {4},
pages = {2152–2158},
abstract = {Carbon nanotubes are promising building blocks for various nanoelectronic components. A highly desirable geometry for such applications is a coil. However, coiled nanotube structures reported so far were inherently defective or had no free ends accessible for contacting. Here we demonstrate the spontaneous self-coiling of single-wall carbon nanotubes into defect-free coils of up to more than 70 turns with identical diameter and chirality, and free ends. We characterize the structure, formation mechanism, and electrical properties of these coils by different microscopies, molecular dynamics simulations, Raman spectroscopy, and electrical and magnetic measurements. The coils are highly conductive, as expected for defect-free carbon nanotubes, but adjacent nanotube segments in the coil are more highly coupled than in regular bundles of single-wall carbon nanotubes, owing to their perfect crystal momentum matching, which enables tunneling between the turns. Although this behavior does not yet enable the performance of these nanotube coils as inductive devices, it does point a clear path for their realization. Hence, this study represents a major step toward the production of many different nanotube coil devices, including inductors, electromagnets, transformers, and dynamos.},
keywords = {CNT, Coils, Molecular Dynamics, Synthesis, TEM},
pubstate = {published},
tppubtype = {article}
}

Gustavo Brunetto Sehmus Ozden, N. S. Karthiselva
Controlled 3D Carbon Nanotube Structures by Plasma Welding Journal Article
In: Advanced Materials Interfaces, vol. 2016, pp. 1500755, 2016.
Abstract | Links | BibTeX | Tags: 3D networks, Carbon Nanotubes, Elasticity, Molecular Dynamics
@article{Ozden2016,
title = {Controlled 3D Carbon Nanotube Structures by Plasma Welding},
author = {Sehmus Ozden, Gustavo Brunetto, N. S. Karthiselva, Douglas S. Galvão, Ajit Roy, Srinivasa R. Bakshi, Chandra S. Tiwary, andPulickel M. Ajayan},
url = {http://onlinelibrary.wiley.com/doi/10.1002/admi.201500755/abstract?campaign=wolearlyview},
doi = {10.1002/admi.201500755},
year = {2016},
date = {2016-03-17},
journal = {Advanced Materials Interfaces},
volume = {2016},
pages = {1500755},
abstract = {3D interconnected carbon nanotubes (CNTs) are synthesized using an industrially scalable spark plasma technique. At high electric field and elevated temperature under sufficient stress the nanotubes are welded together to form a solid block. The detailed spectroscopic and microscopic analyses show successful welding of the CNTs and formation of interconnected networks. The mechanical characteristics of the 3D CNT block show a high stiffness and yield strength. A full atomistic molecular dynamics simulation elucidates the CNT welding mechanism.},
keywords = {3D networks, Carbon Nanotubes, Elasticity, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
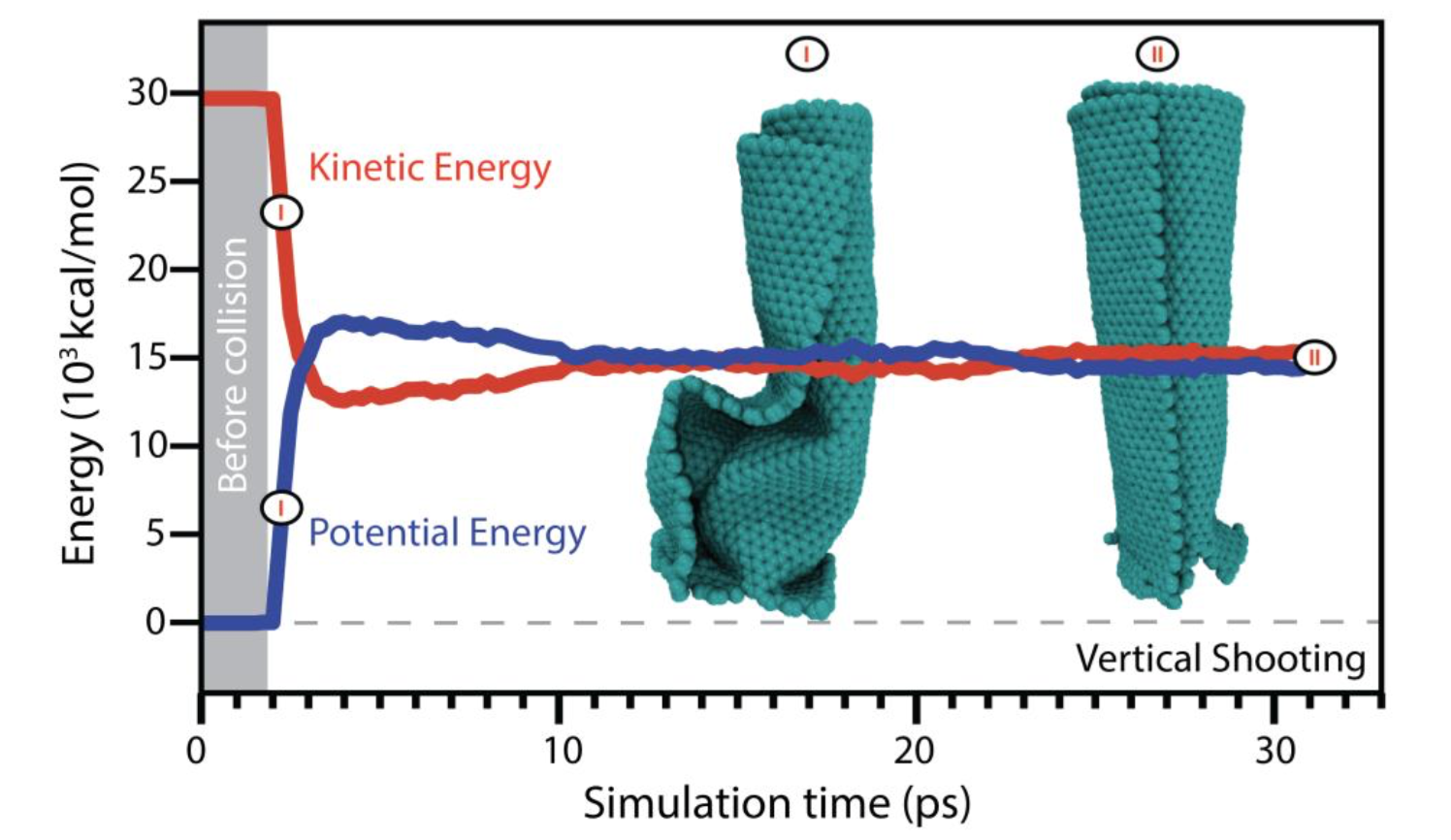
Leonardo Dantas Machado José Moreira de Sousa, Cristiano Francisco Woellner; Galvao, Douglas S.
Carbon Nanoscrolls at High Impacts: A Molecular Dynamics Investigation Journal Article
In: MRS Advances, vol. 2016, 2016.
Abstract | Links | BibTeX | Tags: Impact Molecular Dynamics, nanoscrolls
@article{deSousa2016b,
title = {Carbon Nanoscrolls at High Impacts: A Molecular Dynamics Investigation},
author = {José Moreira de Sousa, Leonardo Dantas Machado, Cristiano Francisco Woellner, Pedro Alves da Silva Autreto and Douglas S. Galvao},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=10242265&fulltextType=RA&fileId=S2059852116002000},
doi = {10.1557/adv.2016.200},
year = {2016},
date = {2016-03-01},
journal = {MRS Advances},
volume = {2016},
abstract = {The behavior of nanostructures under high strain-rate conditions has been object of interest in recent years. For instance, recent experimental investigations showed that at high velocity impacts carbon nanotubes can unzip resulting into graphene nanoribbons. Carbon nanoscrolls (CNS) are among the structures whose high impact behavior has not yet been investigated. CNS are graphene membranes rolled up into papyrus-like structures. Their unique open-ended topology leads to properties not found in close-ended structures, such as nanotubes. Here we report a fully atomistic reactive molecular dynamics study on the behavior of CNS colliding at high velocities against solid targets. Our results show that the velocity and scroll axis orientation are key parameters to determine the resulting formed nanostructures after impact. The relative orientation of the scroll open ends and the substrate is also very important. We observed that for appropriate velocities and orientations, the nanoscrolls can experience large structural deformations and large-scale fractures. We have also observed unscrolling (scrolls going back to planar or quasi-planar graphene membranes), unzip resulting into nanoribbons, and significant reconstructions from breaking and/or formation of new chemical bonds. Another interesting result was that if the CNS impact the substrate with their open ends, for certain velocities, fused scroll walls were observed.},
keywords = {Impact Molecular Dynamics, nanoscrolls},
pubstate = {published},
tppubtype = {article}
}
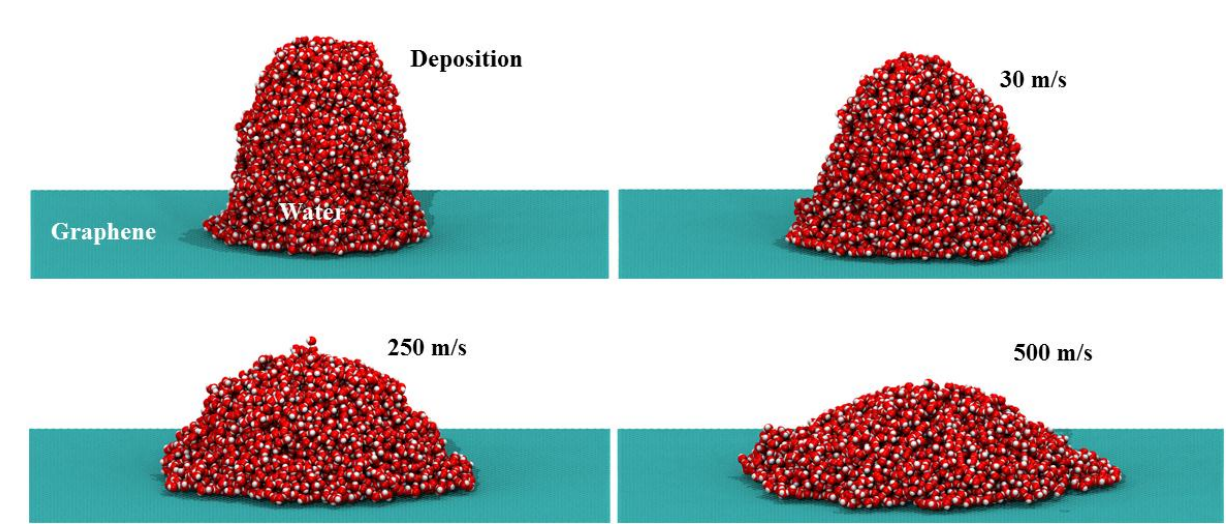
Ygor M. Jaques, Gustavo Brunetto; Galvão, Douglas S.
Nanodroplets Impacting on Graphene Journal Article
In: MRS Advances, vol. 2016, 2016.
Abstract | Links | BibTeX | Tags: Graphene, Impact Molecular Dynamics, nanodroplet
@article{Jaques2016b,
title = {Nanodroplets Impacting on Graphene},
author = {Ygor M. Jaques, Gustavo Brunetto and Douglas S. Galvão},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=10253580&fulltextType=RA&fileId=S2059852116002218},
doi = {DOI: 10.1557/adv.2016.221},
year = {2016},
date = {2016-03-01},
journal = {MRS Advances},
volume = {2016},
abstract = {The unique and remarkable properties of graphene can be exploited as the basis to a wide
range of applications. However, in spite of years of investigations there are some important
graphene properties that are not still fully understood, as for example, its wettability. There are
controversial reported results whether graphene is really hydrophobic or hydrophilic. In order to
address this problem we have carried out classical molecular dynamics simulations of water
nanodroplets shot against graphene surface. Our results show that the contact angle values
between the nanodroplets and graphene surfaces depend on the initial droplet velocity value and
these angles can change from 86º (hydrophobic) to 35º (hydrophilic). Our preliminary results
indicate that the graphene wettability can be dependent on spreading liquid dynamics and which
can explain some of the apparent inconsistencies reported in the literature.},
keywords = {Graphene, Impact Molecular Dynamics, nanodroplet},
pubstate = {published},
tppubtype = {article}
}
range of applications. However, in spite of years of investigations there are some important
graphene properties that are not still fully understood, as for example, its wettability. There are
controversial reported results whether graphene is really hydrophobic or hydrophilic. In order to
address this problem we have carried out classical molecular dynamics simulations of water
nanodroplets shot against graphene surface. Our results show that the contact angle values
between the nanodroplets and graphene surfaces depend on the initial droplet velocity value and
these angles can change from 86º (hydrophobic) to 35º (hydrophilic). Our preliminary results
indicate that the graphene wettability can be dependent on spreading liquid dynamics and which
can explain some of the apparent inconsistencies reported in the literature.
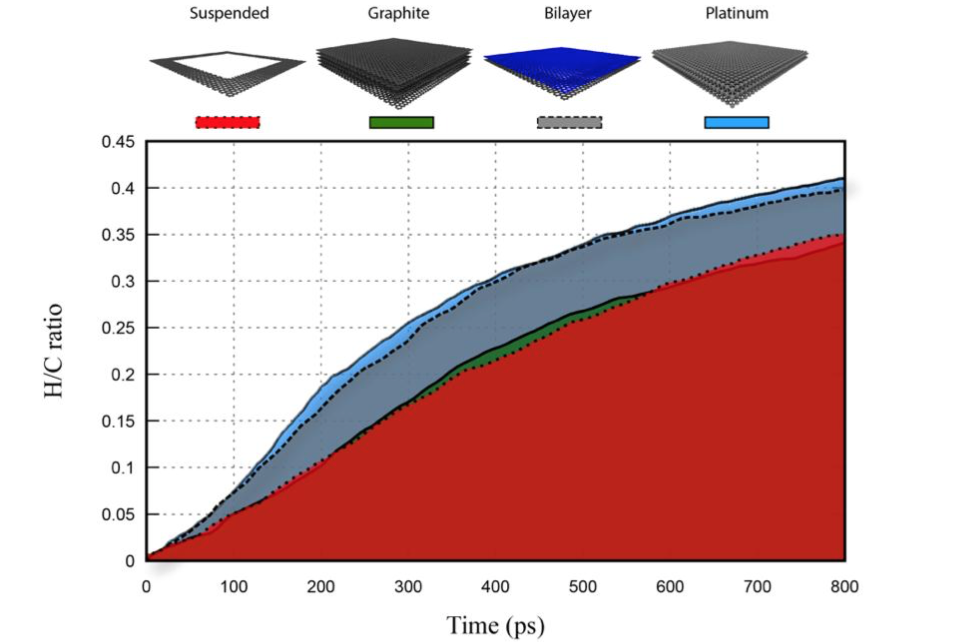
Pedro Alves da Silva Autreto Cristiano Francisco Woellner, Douglas S. Galvao
One Side-Graphene Hydrogenation (Graphone): Substrate Effects Journal Article
In: MRS Advances, vol. 2016, 2016.
Abstract | Links | BibTeX | Tags: Graphane, Graphene, graphone, Molecular Dynamics
@article{Woellner2016b,
title = {One Side-Graphene Hydrogenation (Graphone): Substrate Effects},
author = {Cristiano Francisco Woellner, Pedro Alves da Silva Autreto, Douglas S. Galvao},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=10234793&fulltextType=RA&fileId=S2059852116001961},
doi = {DOI: 10.1557/adv.2016.196},
year = {2016},
date = {2016-03-01},
journal = {MRS Advances},
volume = {2016},
abstract = {Recent studies on graphene hydrogenation processes showed that hydrogenation occurs via island growing domains, however how the substrate can affect the hydrogenation dynamics and/or pattern formation has not been yet properly investigated. In this work we have addressed these issues through fully atomistic reactive molecular dynamics simulations. We investigated the structural and dynamical aspects of the hydrogenation of graphene membranes (one-side hydrogenation, the so called graphone structure) on different substrates (graphene, few-layers graphene, graphite and platinum). Our results also show that the observed hydrogenation rates are very sensitive to the substrate type. For all investigated cases, the largest fraction of hydrogenated carbon atoms was for platinum substrates. Our results also show that a significant number of randomly distributed H clusters are formed during the early stages of the hydrogenation process, regardless of the type of substrate. These results suggest that, similarly to graphane formation, large perfect graphone-like domains are unlikely to be formed. These findings are especially important since experiments have showed that cluster formation influences the electronic transport properties in hydrogenated graphene.
},
keywords = {Graphane, Graphene, graphone, Molecular Dynamics},
pubstate = {published},
tppubtype = {article}
}
Daff, Thomas D; Collins, Sean P; Durekova, Hana; Perim, E; Skaf, Munir S; Galvão, Douglas S; Woo, Tom K
Evaluation of carbon nanoscroll materials for post-combustion CO2 capture Journal Article
In: Carbon, vol. 101, pp. 218–225, 2016.
Abstract | Links | BibTeX | Tags: CO2 capture, Molecular Dynamics, Scrolls
@article{Daff2016,
title = {Evaluation of carbon nanoscroll materials for post-combustion CO2 capture},
author = {Daff, Thomas D and Collins, Sean P and Durekova, Hana and Perim, E and Skaf, Munir S and Galvão, Douglas S and Woo, Tom K},
url = {http://www.sciencedirect.com/science/article/pii/S0008622316300604},
doi = {10.1016/j.carbon.2016.01.072},
year = {2016},
date = {2016-02-11},
journal = {Carbon},
volume = {101},
pages = {218–225},
abstract = {Carbon nanoscrolls are similar to multi-walled carbon nanotubes but constructed from rolled graphene sheets into papyrus-like structures. In this work, molecular simulations are used to evaluate the post-combustion CO2 capture properties of nanoscrolls made of graphene, α-, β-, and γ-graphyne, boron nitride, and three types of carbon nitride. The CO2 uptake capacity, CO2/N2 selectivity and CO2 working capacity were computed with grand canonical Monte Carlo simulations at conditions relevant to post-combustion CO2 capture. The interlayer spacing of the nanoscrolls was optimized for each property and sheet material. For graphene nanoscrolls, the optimal interlayer spacing of 7.3 Å was identified for both the CO2 uptake and selectivity, while for working capacity the optimal interlayer spacing was determined to be 8.6 Å. It was found that the CO2 uptake capacity of the materials correlated to the density of the sheets from which they were formed. Nanoscrolls made from graphene and boron nitride, which have the highest number of atoms per unit area, also showed the highest CO2 uptakes. At 0.15 bar CO2, 313 K, graphene and boron nitride nanoscrolls exhibited exceptional CO2 uptake capacities of 7.7 and 8.2 mmol/g, respectively, while also exhibiting high CO2/N2 selectivities of 135 and 153, respectively. Molecular dynamics simulations were used to examine the adsorption kinetics. The simulations showed that an empty graphene nanoscroll with a roll length of 200 Å could adsorb CO2 into the center of the roll within 10 ns. Materials with pores that can allow CO2 to pass through, such as graphynes, showed much faster adsorption times.},
keywords = {CO2 capture, Molecular Dynamics, Scrolls},
pubstate = {published},
tppubtype = {article}
}

Jaques, Ygor M.; Brunetto, Gustavo; Galvao, Douglas S.
Nanodroplets Impacting on Graphene Online
2016, ((ArXiv preprint)).
Abstract | Links | BibTeX | Tags: Droplet, Graphene, Molecular Dynamics
@online{Jaques2016,
title = {Nanodroplets Impacting on Graphene},
author = {Jaques, Ygor M. and Brunetto, Gustavo and Galvao, Douglas S.},
url = {http://arxiv.org/abs/1602.02013},
year = {2016},
date = {2016-02-05},
abstract = {The unique and remarkable properties of graphene can be exploited as the basis to a wide
range of applications. However, in spite of years of investigations there are some important
graphene properties that are not still fully understood, as for example, its wettability. There are
controversial reported results whether graphene is really hydrophobic or hydrophilic. In order to
address this problem we have carried out classical molecular dynamics simulations of water
nanodroplets shot against graphene surface. Our results show that the contact angle values
between the nanodroplets and graphene surfaces depend on the initial droplet velocity value and
these angles can change from 86º (hydrophobic) to 35º (hydrophilic). Our preliminary results
indicate that the graphene wettability can be dependent on spreading liquid dynamics and which
can explain some of the apparent inconsistencies reported in the literature.},
note = {(ArXiv preprint)},
keywords = {Droplet, Graphene, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}
range of applications. However, in spite of years of investigations there are some important
graphene properties that are not still fully understood, as for example, its wettability. There are
controversial reported results whether graphene is really hydrophobic or hydrophilic. In order to
address this problem we have carried out classical molecular dynamics simulations of water
nanodroplets shot against graphene surface. Our results show that the contact angle values
between the nanodroplets and graphene surfaces depend on the initial droplet velocity value and
these angles can change from 86º (hydrophobic) to 35º (hydrophilic). Our preliminary results
indicate that the graphene wettability can be dependent on spreading liquid dynamics and which
can explain some of the apparent inconsistencies reported in the literature.
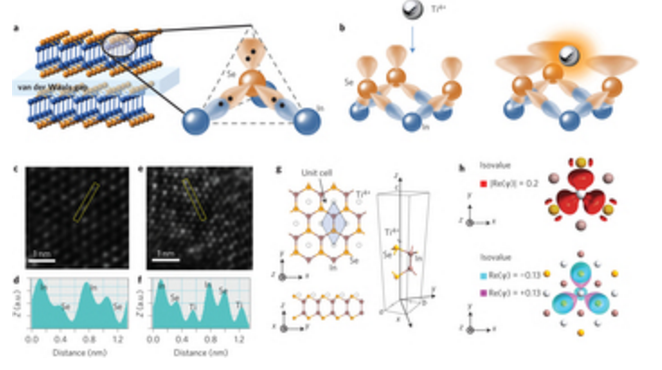
Xifan Wang Sidong Lei, Bo Li
Surface functionalization of two-dimensional metal chalcogenides by Lewis acid–base chemistry Journal Article
In: Nature Nanotechnology, vol. 11, pp. 465–471, 2016.
Abstract | Links | BibTeX | Tags: Chalcogenides, Modelling, Synthesis, top20
@article{Lei2016,
title = {Surface functionalization of two-dimensional metal chalcogenides by Lewis acid–base chemistry},
author = {Sidong Lei, Xifan Wang, Bo Li, Jiahao Kang, Yongmin He, Antony George, Liehui Ge, Yongji Gong, Pei Dong, Zehua Jin, Gustavo Brunetto, Weibing Chen, Zuan-Tao Lin, Robert Baines, Douglas S. Galvão, Jun Lou, Enrique Barrera, Kaustav Banerjee, Robert Vajtai & Pulickel Ajayan},
url = {http://www.nature.com/nnano/journal/vaop/ncurrent/full/nnano.2015.323.html},
doi = {10.1038/nnano.2015.323},
year = {2016},
date = {2016-02-01},
journal = {Nature Nanotechnology},
volume = {11},
pages = {465–471},
abstract = {Precise control of the electronic surface states of two-dimensional (2D) materials could improve their versatility and widen their applicability in electronics and sensing. To this end, chemical surface functionalization has been used to adjust the electronic properties of 2D materials. So far, however, chemical functionalization has relied on lattice defects and physisorption methods that inevitably modify the topological characteristics of the atomic layers. Here we make use of the lone pair electrons found in most of 2D metal chalcogenides and report a functionalization method via a Lewis acid–base reaction that does not alter the host structure. Atomic layers of n-type InSe react with Ti4+ to form planar p-type [Ti4+n(InSe)] coordination complexes. Using this strategy, we fabricate planar p–n junctions on 2D InSe with improved rectification and photovoltaic properties, without requiring heterostructure growth procedures or device fabrication processes. We also show that this functionalization approach works with other Lewis acids (such as B3+, Al3+ and Sn4+) and can be applied to other 2D materials (for example MoS2, MoSe2). Finally, we show that it is possible to use Lewis acid–base chemistry as a bridge to connect molecules to 2D atomic layers and fabricate a proof-of-principle dye-sensitized photosensing device.
},
keywords = {Chalcogenides, Modelling, Synthesis, top20},
pubstate = {published},
tppubtype = {article}
}

de Sousa, Jose Moreira; Machado, Leonardo Dantas; Woellner, Cristiano Francisco; Autreto, Pedro Alves da Silva; Galvao, Douglas S
Carbon Nanoscrolls at High Impacts: A Molecular Dynamics Investigation Online
2016, ((ArXiv Preprint)).
Abstract | Links | BibTeX | Tags: Ballistic Impact, Molecular Dynamics, Scrolls
@online{deSousa2016b,
title = {Carbon Nanoscrolls at High Impacts: A Molecular Dynamics Investigation},
author = {de Sousa, Jose Moreira and Machado, Leonardo Dantas and Woellner, Cristiano Francisco and Autreto, Pedro Alves da Silva and Galvao, Douglas S},
url = {http://arxiv.org/abs/1601.04875},
year = {2016},
date = {2016-01-19},
abstract = {The behavior of nanostructures under high strain-rate conditions has been object of interest in recent years. For instance, recent experimental investigations showed that at high velocity impacts carbon nanotubes can unzip resulting into graphene nanoribbons. Carbon nanoscrolls (CNS) are among the structures whose high impact behavior has not yet been investigated. CNS are graphene membranes rolled up into papyrus-like structures. Their unique open-ended topology leads to properties not found in close-ended structures, such as nanotubes. Here we report a fully atomistic reactive molecular dynamics study on the behavior of CNS colliding at high velocities against solid targets. Our results show that the velocity and scroll axis orientation are key parameters to determine the resulting formed nanostructures after impact. The relative orientation of the scroll open ends and the substrate is also very important. We observed that for appropriate velocities and orientations, the nanoscrolls can experience large structural deformations and large-scale fractures. We have also observed unscrolling (scrolls going back to planar or quasi-planar graphene membranes), unzip resulting into nanoribbons, and significant reconstructions from breaking and/or formation of new chemical bonds. Another interesting result was that if the CNS impact the substrate with their open ends, for certain velocities, fused scroll walls were observed.},
note = {(ArXiv Preprint)},
keywords = {Ballistic Impact, Molecular Dynamics, Scrolls},
pubstate = {published},
tppubtype = {online}
}
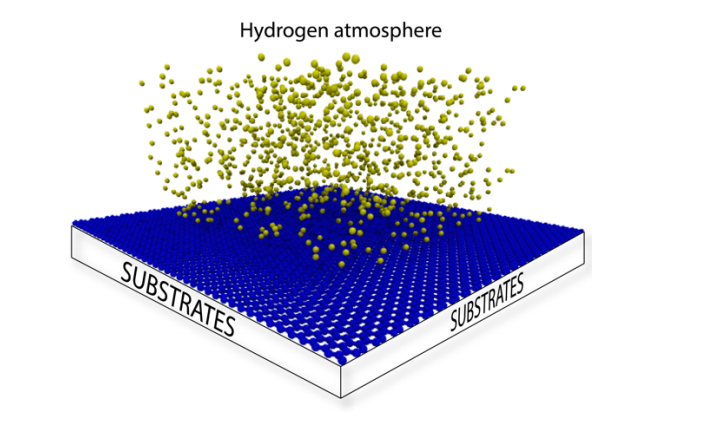
Woellner, Cristiano Francisco; Autreto, Pedro Alves da Silva; Galvao, Douglas S
One Side-Graphene Hydrogenation (Graphone): Substrate Effects Online
2016, visited: 18.01.2016, ((ArXiv preprint)).
Abstract | Links | BibTeX | Tags: Graphane, Graphene, graphone, Molecular Dynamics
@online{Woellner2016,
title = {One Side-Graphene Hydrogenation (Graphone): Substrate Effects},
author = {Woellner, Cristiano Francisco and Autreto, Pedro Alves da Silva and Galvao, Douglas S},
url = {http://arxiv.org/abs/1601.04484},
year = {2016},
date = {2016-01-18},
urldate = {2016-01-18},
abstract = {Recent studies on graphene hydrogenation processes showed that hydrogenation occurs
via island growing domains, however how the substrate can affect the hydrogenation dynamics
and/or pattern formation has not been yet properly investigated. In this work we have addressed
these issues through fully atomistic reactive molecular dynamics simulations. We investigated
the structural and dynamical aspects of the hydrogenation of graphene membranes (one-side
hydrogenation, the so called graphone structure) on different substrates (graphene, few-layers
graphene, graphite and platinum). Our results also show that the observed hydrogenation rates
are very sensitive to the substrate type. For all investigated cases, the largest fraction of
hydrogenated carbon atoms was for platinum substrates. Our results also show that a significant
number of randomly distributed H clusters are formed during the early stages of the
hydrogenation process, regardless of the type of substrate and temperature. These results suggest
that, similarly to graphane formation, large perfect graphone-like domains are unlikely to be
formed. These findings are especially important since experiments have showed that cluster
formation influences the electronic transport properties in hydrogenated graphene.},
note = {(ArXiv preprint)},
keywords = {Graphane, Graphene, graphone, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}
via island growing domains, however how the substrate can affect the hydrogenation dynamics
and/or pattern formation has not been yet properly investigated. In this work we have addressed
these issues through fully atomistic reactive molecular dynamics simulations. We investigated
the structural and dynamical aspects of the hydrogenation of graphene membranes (one-side
hydrogenation, the so called graphone structure) on different substrates (graphene, few-layers
graphene, graphite and platinum). Our results also show that the observed hydrogenation rates
are very sensitive to the substrate type. For all investigated cases, the largest fraction of
hydrogenated carbon atoms was for platinum substrates. Our results also show that a significant
number of randomly distributed H clusters are formed during the early stages of the
hydrogenation process, regardless of the type of substrate and temperature. These results suggest
that, similarly to graphane formation, large perfect graphone-like domains are unlikely to be
formed. These findings are especially important since experiments have showed that cluster
formation influences the electronic transport properties in hydrogenated graphene.
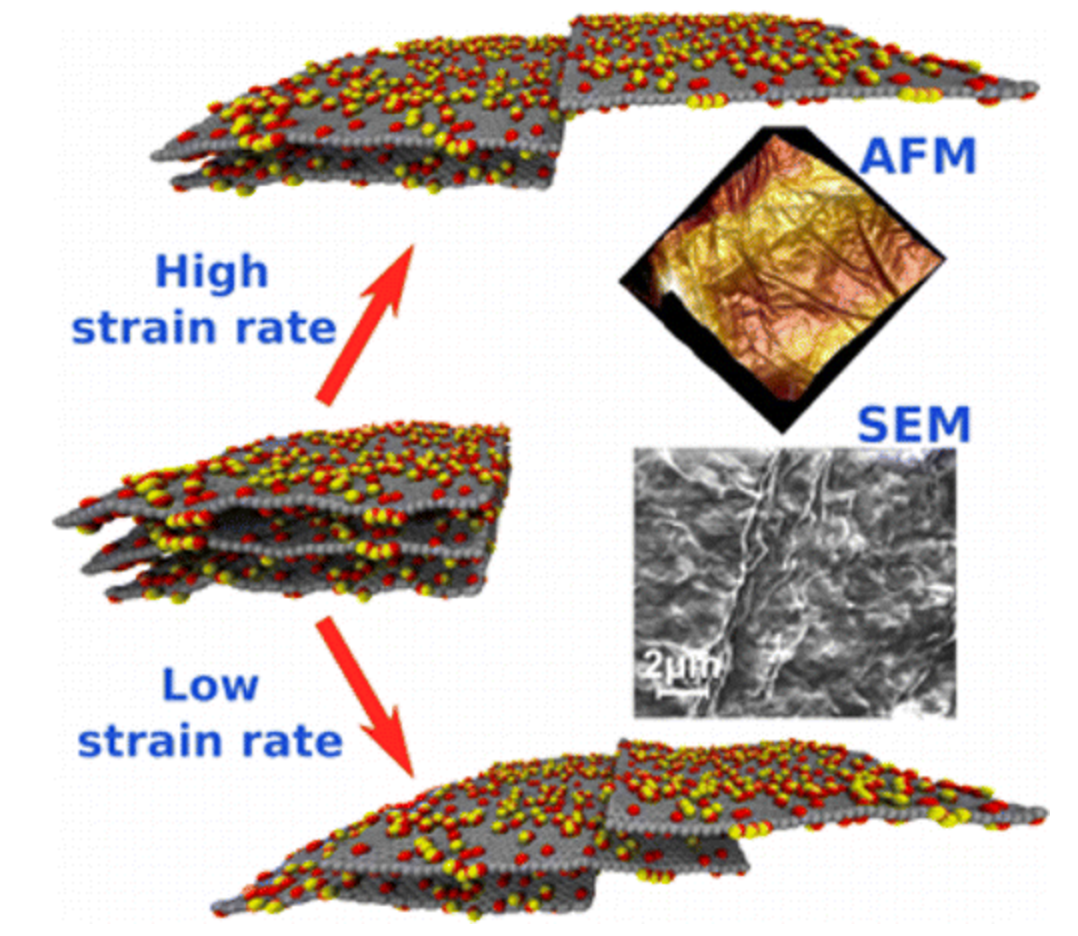
Vinod, Soumya; Tiwary, Chandra Sekhar; Machado, Leonardo Dantas; Ozden, Sehmus; Shaw, Preston; Cho, Juny; Vajtai, Robert; Galvao, Douglas Soares; Ajayan, Pulickel M
Strain Rate Dependent Shear Plasticity in Graphite Oxide Journal Article
In: Nano Letters, vol. 16, no. 2, pp. 1127–1131, 2016.
Abstract | Links | BibTeX | Tags: graphene oxide, Molecular Dynamics, plasticity
@article{Vinod2016,
title = {Strain Rate Dependent Shear Plasticity in Graphite Oxide},
author = {Vinod, Soumya and Tiwary, Chandra Sekhar and Machado, Leonardo Dantas and Ozden, Sehmus and Shaw, Preston and Cho, Juny and Vajtai, Robert and Galvao, Douglas Soares and Ajayan, Pulickel M},
url = {http://pubs.acs.org/doi/abs/10.1021/acs.nanolett.5b04346},
doi = {10.1021/acs.nanolett.5b04346},
year = {2016},
date = {2016-01-16},
journal = {Nano Letters},
volume = {16},
number = {2},
pages = {1127–1131},
abstract = {Graphene oxide film is made of stacked graphene layers with chemical functionalities, and we report that plasticity in the film can be engineered by strain rate tuning. The deformation behavior and plasticity of such functionalized layered systems is dominated by shear slip between individual layers and interaction between functional groups. Stress–strain behavior and theoretical models suggest that the deformation is strongly strain rate dependent and undergoes brittle to ductile transition with decreasing strain rate.},
keywords = {graphene oxide, Molecular Dynamics, plasticity},
pubstate = {published},
tppubtype = {article}
}
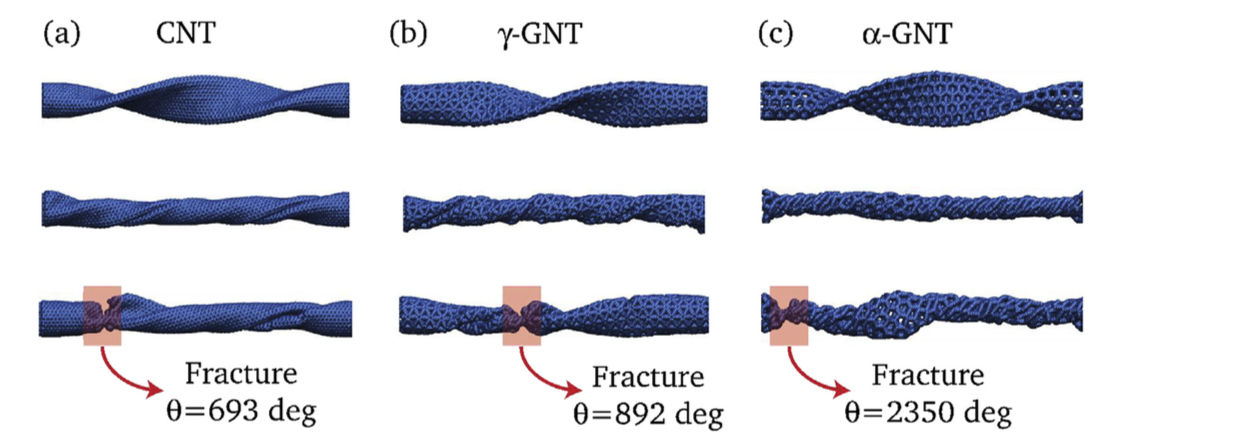
G. Brunetto J.M. de Sousa, V. R. Coluci
Torsional “superplasticity” of graphyne nanotubes Journal Article
In: Carbon, vol. 96, pp. 14-19, 2016.
Abstract | Links | BibTeX | Tags: Fracture, Graphynes, Mechanical Properties, Nanotubes
@article{deSousa2016,
title = {Torsional “superplasticity” of graphyne nanotubes},
author = {J.M. de Sousa, G. Brunetto, V.R. Coluci, D.S. Galvao },
url = {http://www.sciencedirect.com/science/article/pii/S000862231530258X},
doi = { http://dx.doi.org/10.1016/j.carbon.2015.09.039},
year = {2016},
date = {2016-01-01},
journal = {Carbon},
volume = {96},
pages = {14-19},
abstract = {Graphyne is a planar two-dimensional carbon allotrope formed by atoms in sp, sp2, and sp3 hybridized states. Topologically graphyne nanotubes (GNTs) can be considered as cylindrically rolled up graphyne sheets, similarly as carbon nanotubes (CNTs) can be considered rolled up graphene sheets. Due to the presence of single, double, and triple bonds, GNTs exhibit porous sidewalls that can be exploited in many diverse applications. In this work, we investigated the mechanical behavior of GNTs under torsional strains through reactive molecular dynamics simulations. Our results show that GNTs are more flexible than CNTs and exhibit “superplasticit”, with fracture angles that are up to 35 times higher than the ones reported to CNTs. This GNT “superplastic” behavior can be explained in terms of irreversible recon- struction processes (mainly associated with the triple bonds) that occur during torsional strains.},
keywords = {Fracture, Graphynes, Mechanical Properties, Nanotubes},
pubstate = {published},
tppubtype = {article}
}

Dong, Pei; Chipara, Alin Cristian; Loya, Phillip; Yang, Yingchao; Ge, Liehui; Lei, Sidong; Li, Bo; Brunetto, Gustavo; Machado, Leonardo Dantas; Hong, Liang; others,
A Solid-liquid Self-adaptive Polymeric Composite Journal Article
In: ACS Applied Materials & Interfaces, vol. 8, no. 3, pp. 2142–2147, 2016.
Abstract | Links | BibTeX | Tags: Adhesives, Modelling, Polymers
@article{Dong2016,
title = {A Solid-liquid Self-adaptive Polymeric Composite},
author = {Dong, Pei and Chipara, Alin Cristian and Loya, Phillip and Yang, Yingchao and Ge, Liehui and Lei, Sidong and Li, Bo and Brunetto, Gustavo and Machado, Leonardo Dantas and Hong, Liang and others},
url = {http://pubs.acs.org/doi/abs/10.1021/acsami.5b10667},
doi = {10.1021/acsami.5b10667},
year = {2016},
date = {2016-01-01},
journal = {ACS Applied Materials & Interfaces},
volume = {8},
number = {3},
pages = {2142–2147},
abstract = {A solid–liquid self-adaptive composite (SAC) is synthesized using a simple mixing–evaporation protocol, with poly(dimethylsiloxane) (PDMS) and poly(vinylidene fluoride) (PVDF) as active constituents. SAC exists as a porous solid containing a near equivalent distribution of the solid (PVDF)–liquid (PDMS) phases, with the liquid encapsulated and stabilized within a continuous solid network percolating throughout the structure. The pores, liquid, and solid phases form a complex hierarchical structure, which offers both mechanical robustness and a significant structural adaptability under external forces. SAC exhibits attractive self-healing properties during tension, and demonstrates reversible self-stiffening properties under compression with a maximum of 7-fold increase seen in the storage modulus. In a comparison to existing self-healing and self-stiffening materials, SAC offers distinct advantages in the ease of fabrication, high achievable storage modulus, and reversibility. Such materials could provide a new class of adaptive materials system with multifunctionality, tunability, and scale-up potentials.
},
keywords = {Adhesives, Modelling, Polymers},
pubstate = {published},
tppubtype = {article}
}
2015
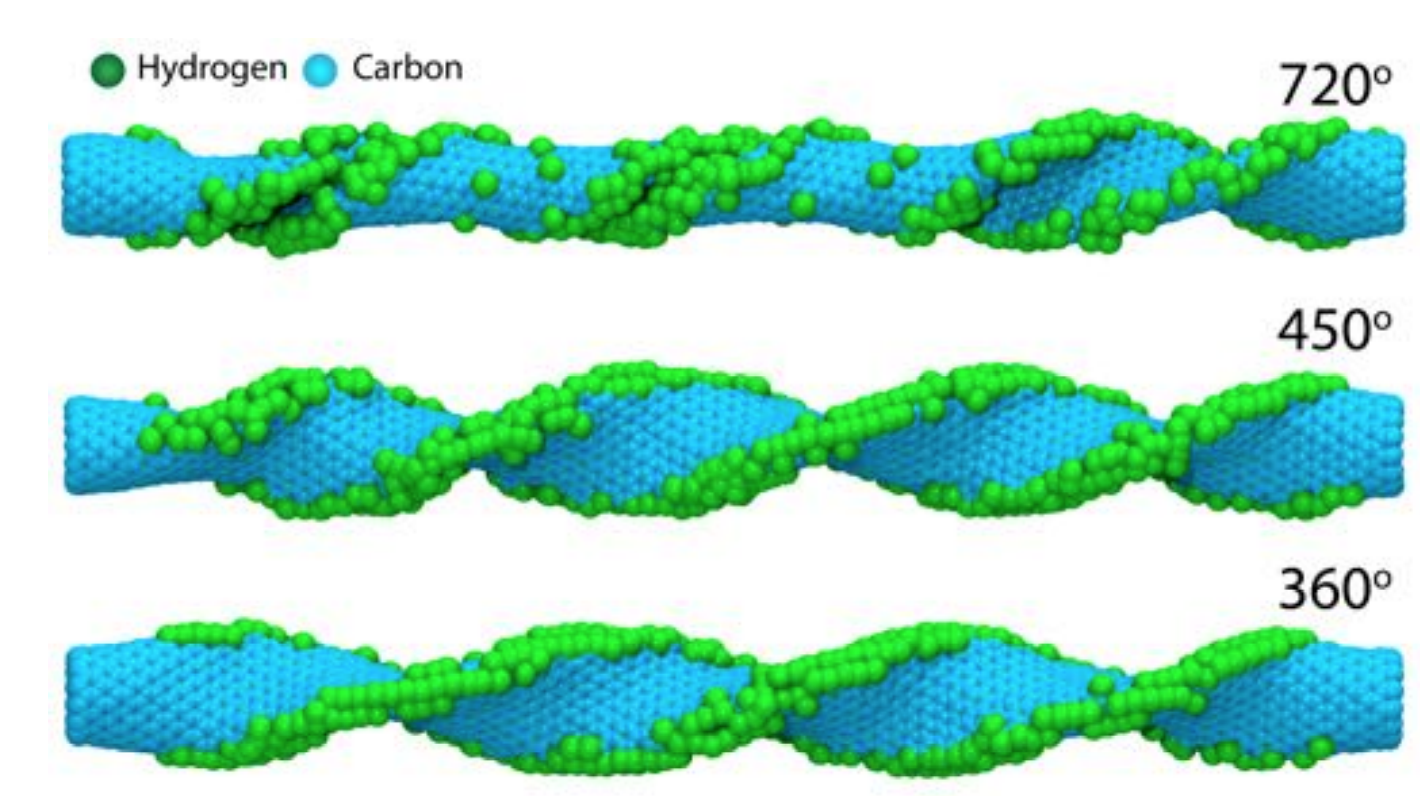
de Sousa, Jose M.; Autreto, Pedro A. S.; Galvao, Douglas S.
Hydrogenation Dynamics of Twisted Carbon Nanotubes Online
2015, (ArXiv preprint).
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, Hydrogenation, Mechanical Properties, Molecular Dynamics
@online{deSousa2015,
title = {Hydrogenation Dynamics of Twisted Carbon Nanotubes},
author = {Jose M. de Sousa and Pedro A. S. Autreto and Douglas S. Galvao},
url = {http://arxiv.org/abs/1510.00265},
year = {2015},
date = {2015-10-01},
abstract = {Carbon Nanotubes (CNTs) are one of the most important materials in nanotechnology. In some of their technological applications (electromechanical oscillators and mechanical actuators for artificial muscles, for instance), it is necessary to subject them to large deformations. Although this frequently happens in air, there are only few studies about the interaction of deformed CNTs with the atmosphere and the dynamics of these processes has not yet been addressed. In this work, we have investigated, through fully atomistic reactive molecular dynamics simulations, the process of hydrogenation of highly twisted CNTs. Our results show that hydrogenation effective ratio is directly related to the tube twist angle values and can lead to twisted tube fractures with well defined patterns (unzip-like). Our results also show that these fracture processes can be exploited to controllably produce graphene nanoribbons.},
note = {ArXiv preprint},
keywords = {Carbon Nanotubes, Hydrogenation, Mechanical Properties, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}
Gustavo Brunetto Jose M. de Sousa, Vitor R. Coluci
Torsional "Superplasticity" of Graphyne Nanotubes Online
2015, (ArXiv reprint of Torsional "Superplasticity" of Graphyne Nanotubes, published in Carbon 96, 14 (2016).).
Abstract | Links | BibTeX | Tags: Allotropes, Graphynes, Mechanical Properties, Nanotubes
@online{deSousa2015b,
title = {Torsional "Superplasticity" of Graphyne Nanotubes},
author = {Jose M. de Sousa, Gustavo Brunetto, Vitor R. Coluci, Douglas S. Galvao},
url = {http://arxiv.org/abs/1509.08746},
year = {2015},
date = {2015-09-29},
abstract = {Graphyne is a planar two-dimensional carbon allotrope formed by atoms in sp, sp2, and sp3 hybridized states. Topologically graphyne nanotubes (GNTs) can be considered as cylindrically rolled up graphyne sheets, similarly as carbon nanotubes (CNTs) can be considered rolled up graphene sheets. Due to the presence of single, double, and triple bonds, GNTs exhibit porous sidewalls that can be exploited in many diverse applications. In this work, we investigated the mechanical behavior of GNTs under torsional strains through reactive molecular dynamics simulations. Our results show that GNTs are more flexible than CNTs and exhibit 'superplasticity', with fracture angles that are up to 35 times higher than the ones reported to CNTs. This GNT 'superplastic' behavior can be explained in terms of irreversible reconstruction processes (mainly associated with the triple bonds) that occur during torsional strains.},
note = {ArXiv reprint of Torsional "Superplasticity" of Graphyne Nanotubes, published in Carbon 96, 14 (2016).},
keywords = {Allotropes, Graphynes, Mechanical Properties, Nanotubes},
pubstate = {published},
tppubtype = {online}
}
S Fang ZF Liu, FA Moura
Hierarchically buckled sheath-core fibers for superelastic electronics, sensors, and muscles Journal Article
In: Science, vol. 349, no. 6246, pp. 404-404, 2015.
Abstract | Links | BibTeX | Tags: Carbon Nanotube Forests, Finite Elements, Superelastic, top20
@article{Liu2015,
title = {Hierarchically buckled sheath-core fibers for superelastic electronics, sensors, and muscles},
author = {ZF Liu, S Fang, FA Moura, JN Ding, N Jiang, J Di, M Zhang, X Lepró, DS Galvão, CS Haines, NY Yuan, SG Yin, DW Lee, R Wang, HY Wang, W Lv, C Dong, RC Zhang, MJ Chen, Q Yin, YT Chong, R Zhang, X Wang, MD Lima, R Ovalle-Robles, D Qian, H Lu, RH Baughman},
url = {http://www.sciencemag.org/content/349/6246/400.full.pdf},
doi = {10.1126/science.aaa7952},
year = {2015},
date = {2015-07-24},
journal = {Science},
volume = {349},
number = {6246},
pages = {404-404},
abstract = {Superelastic conducting fibers with improved properties and functionalities are needed
for diverse applications. Here we report the fabrication of highly stretchable (up to 1320%)
sheath-core conducting fibers created by wrapping carbon nanotube sheets oriented in
the fiber direction on stretched rubber fiber cores. The resulting structure exhibited
distinct short- and long-period sheath buckling that occurred reversibly out of phase
in the axial and belt directions, enabling a resistance change of less than 5% for a
1000% stretch. By including other rubber and carbon nanotube sheath layers, we
demonstrated strain sensors generating an 860% capacitance change and electrically
powered torsional muscles operating reversibly by a coupled tension-to-torsion
actuation mechanism. Using theory, we quantitatively explain the complementary effects
of an increase in muscle length and a large positive Poisson’s ratio on torsional actuation
and electronic properties.},
keywords = {Carbon Nanotube Forests, Finite Elements, Superelastic, top20},
pubstate = {published},
tppubtype = {article}
}
for diverse applications. Here we report the fabrication of highly stretchable (up to 1320%)
sheath-core conducting fibers created by wrapping carbon nanotube sheets oriented in
the fiber direction on stretched rubber fiber cores. The resulting structure exhibited
distinct short- and long-period sheath buckling that occurred reversibly out of phase
in the axial and belt directions, enabling a resistance change of less than 5% for a
1000% stretch. By including other rubber and carbon nanotube sheath layers, we
demonstrated strain sensors generating an 860% capacitance change and electrically
powered torsional muscles operating reversibly by a coupled tension-to-torsion
actuation mechanism. Using theory, we quantitatively explain the complementary effects
of an increase in muscle length and a large positive Poisson’s ratio on torsional actuation
and electronic properties.
Chandra Sekhar Tiwary Dibyendu Chakravarty, Leonardo Dantas Machado
Zirconia-Nanoparticle-Reinforced Morphology-Engineered Graphene-Based Foams Journal Article
In: Advanced Materials, vol. 27, no. 31, pp. 4534–4543, 2015.
Abstract | Links | BibTeX | Tags: Electronic Structure, Mechanical Properties, Mole, Molecular Dynamics, Nanoparticles, Zirconia
@article{Chakravarty2015,
title = {Zirconia-Nanoparticle-Reinforced Morphology-Engineered Graphene-Based Foams},
author = { Dibyendu Chakravarty , Chandra Sekhar Tiwary , Leonardo Dantas Machado ,
Gustavo Brunetto , Soumya Vinod , Ram Manohar Yadav , Douglas S. Galvao ,
Shrikant V. Joshi , Govindan Sundararajan, Pulickel M. Ajayan },
url = {http://onlinelibrary.wiley.com/doi/10.1002/adma.201502409/full},
doi = {10.1002/adma.201502409},
year = {2015},
date = {2015-07-15},
journal = {Advanced Materials},
volume = {27},
number = {31},
pages = {4534–4543},
abstract = {The morphology of graphene-based foams can be engineered by reinforcing them with nanocrystalline zirconia, thus improving their oil-adsorption capacity; This can be observed experimentally and explained theoretically. Low zirconia fractions yield flaky microstructures where zirconia nanoparticles arrest propagating cracks. Higher zirconia concentrations possess a mesh-like interconnected structure where the degree of coiling is dependant on the local zirconia content.},
keywords = {Electronic Structure, Mechanical Properties, Mole, Molecular Dynamics, Nanoparticles, Zirconia},
pubstate = {published},
tppubtype = {article}
}
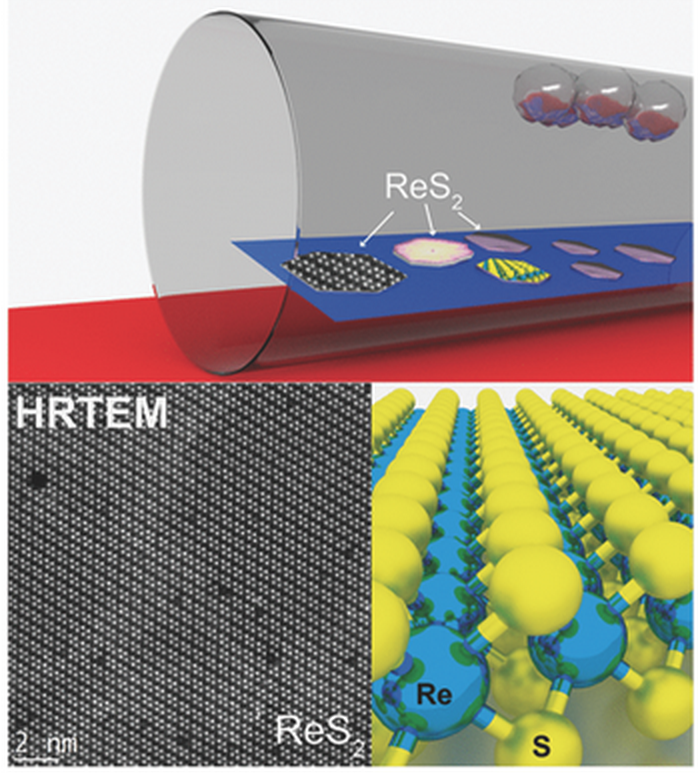
Yongji Gong Kunttal Keyshar, Gonglan Ye
Chemical Vapor Deposition of Monolayer Rhenium Disulfide (ReS2) Journal Article
In: Advanced Materials, vol. 27, no. 31, pp. 4640–4648, 2015.
Abstract | Links | BibTeX | Tags: Electronic Structure, Rhenium Disulfide, Synthesis
@article{Keyshar2015,
title = {Chemical Vapor Deposition of Monolayer Rhenium Disulfide (ReS2)},
author = {Kunttal Keyshar , Yongji Gong , Gonglan Ye , Gustavo Brunetto , Wu Zhou ,
Daniel P. Cole , Ken Hackenberg , Yongmin He , Leonardo Machado , Mohamad Kabbani ,
Amelia H. C. Hart , Bo Li , Douglas S. Galvao , Antony George , Robert Vajtai ,
Chandra Sekhar Tiwary , Pulickel M. Ajayan},
url = {http://onlinelibrary.wiley.com/doi/10.1002/adma.201501795/full},
doi = {10.1002/adma.201501795},
year = {2015},
date = {2015-07-03},
journal = {Advanced Materials},
volume = {27},
number = {31},
pages = {4640–4648},
abstract = {The direct synthesis of monolayer and multilayer ReS2 by chemical vapor deposition at a low temperature of 450 °C is reported. Detailed characterization of this material is performed using various spectroscopy and microscopy methods. Furthermore initial field-effect transistor characteristics are evaluated, which highlight the potential in being used as an n-type semiconductor.},
keywords = {Electronic Structure, Rhenium Disulfide, Synthesis},
pubstate = {published},
tppubtype = {article}
}
Andrei V Alaferdov Victor A Ermakov, Alfredo R Vaz
Burning Graphene Layer-by-Layer Journal Article
In: Nature Scientific Reports, vol. 5, pp. 11546, 2015.
Abstract | Links | BibTeX | Tags: Burning, Graphene, Molecular Dynamics, TEM
@article{Ermakov2015,
title = {Burning Graphene Layer-by-Layer},
author = {Victor A Ermakov, Andrei V Alaferdov, Alfredo R Vaz, Eric Perim, Pedro AS Autreto, Ricardo Paupitz, Douglas S Galvao, Stanislav A Moshkalev},
url = {http://www.nature.com/articles/srep11546?WT.ec_id=SREP-639-20150630},
doi = {10.1038/srep11546},
year = {2015},
date = {2015-06-23},
journal = {Nature Scientific Reports},
volume = {5},
pages = {11546},
abstract = {Graphene, in single layer or multi-layer forms, holds great promise for future electronics and high-temperature applications. Resistance to oxidation, an important property for high-temperature applications, has not yet been extensively investigated. Controlled thinning of multi-layer graphene (MLG), e.g., by plasma or laser processing is another challenge, since the existing methods produce non-uniform thinning or introduce undesirable defects in the basal plane. We report here that heating to extremely high temperatures (exceeding 2000 K) and controllable layer-by-layer burning (thinning) can be achieved by low-power laser processing of suspended high-quality MLG in air in “cold-wall” reactor configuration. In contrast, localized laser heating of supported samples results in non-uniform graphene burning at much higher rates. Fully atomistic molecular dynamics simulations were also performed to reveal details of oxidation mechanisms leading to uniform layer-by-layer graphene gasification. The extraordinary resistance of MLG to oxidation paves the way to novel high-temperature applications as continuum light source or scaffolding material.},
keywords = {Burning, Graphene, Molecular Dynamics, TEM},
pubstate = {published},
tppubtype = {article}
}

Wesller G Schmidt Abraham G Cano-Marquez, Jenaina Ribeiro-Soares
Enhanced Mechanical Stability of Gold Nanotips through Carbon Nanocone Encapsulation Journal Article
In: Nature Scientific Reports, vol. 5, pp. 10408, 2015.
Abstract | Links | BibTeX | Tags: Gold Nanotips, Molecular Dynamics, Nanocones
@article{Cano-Marquez2015,
title = {Enhanced Mechanical Stability of Gold Nanotips through Carbon Nanocone Encapsulation},
author = {Abraham G Cano-Marquez, Wesller G Schmidt, Jenaina Ribeiro-Soares, Luiz Gustavo Cançado, Wagner N Rodrigues, Adelina P Santos, Clascidia A Furtado, Pedro AS Autreto, Ricardo Paupitz, Douglas S Galvão, Ado Jorio},
url = {http://www.nature.com/articles/srep10408},
doi = {10.1038/srep10408},
year = {2015},
date = {2015-06-17},
journal = {Nature Scientific Reports},
volume = {5},
pages = {10408},
abstract = {Gold is a noble metal that, in comparison with silver and copper, has the advantage of corrosion resistance. Despite its high conductivity, chemical stability and biocompatibility, gold exhibits high plasticity, which limits its applications in some nanodevices. Here, we report an experimental and theoretical study on how to attain enhanced mechanical stability of gold nanotips. The gold tips were fabricated by chemical etching and further encapsulated with carbon nanocones via nanomanipulation. Atomic force microscopy experiments were carried out to test their mechanical stability. Molecular dynamics simulations show that the encapsulated nanocone changes the strain release mechanisms at the nanoscale by blocking gold atomic sliding, redistributing the strain along the whole nanostructure. The carbon nanocones are conducting and can induce magnetism, thus opening new avenues on the exploitation of transport, mechanical and magnetic properties of gold covered by sp2 carbon at the nanoscale.},
keywords = {Gold Nanotips, Molecular Dynamics, Nanocones},
pubstate = {published},
tppubtype = {article}
}
Chandra Sekhar Tiwary Mohamad A Kabbani, Pedro AS Autreto
Ambient solid-state mechano-chemical reactions between functionalized carbon nanotubes Journal Article
In: Nature Communications, vol. 6, pp. 7291, 2015.
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, Chemical Reactions, Electronic Structure, Molecular Dynamics, top20
@article{Kabbani2015,
title = {Ambient solid-state mechano-chemical reactions between functionalized carbon nanotubes},
author = {Mohamad A Kabbani, Chandra Sekhar Tiwary, Pedro AS Autreto, Gustavo Brunetto, Anirban Som, KR Krishnadas, Sehmus Ozden, Ken P Hackenberg, Yongi Gong, Douglas S Galvao, Robert Vajtai, Ahmad T Kabbani, Thalappil Pradeep, Pulickel M Ajayan},
url = {http://www.nature.com/ncomms/2015/150615/ncomms8291/full/ncomms8291.html},
doi = {10.1038/ncomms8291},
year = {2015},
date = {2015-06-15},
journal = {Nature Communications},
volume = {6},
pages = {7291},
abstract = {Carbon nanotubes can be chemically modified by attaching various functionalities to their surfaces, although harsh chemical treatments can lead to their break-up into graphene nanostructures. On the other hand, direct coupling between functionalities bound on individual nanotubes could lead to, as yet unexplored, spontaneous chemical reactions. Here we report an ambient mechano-chemical reaction between two varieties of nanotubes, carrying predominantly carboxyl and hydroxyl functionalities, respectively, facilitated by simple mechanical grinding of the reactants. The purely solid-state reaction between the chemically differentiated nanotube species produces condensation products and unzipping of nanotubes due to local energy release, as confirmed by spectroscopic measurements, thermal analysis and molecular dynamic simulations.},
keywords = {Carbon Nanotubes, Chemical Reactions, Electronic Structure, Molecular Dynamics, top20},
pubstate = {published},
tppubtype = {article}
}
Acrísio L Aguiar Nadia Ferreira Andrade, Yoong Ahm Kim
Linear Carbon Chains Under High Pressure Conditions Journal Article
In: The Journal of Physical Chemistry C, vol. 119, no. 19, pp. 10669–10676, 2015.
Abstract | Links | BibTeX | Tags: Atomic Chains, Carbon Nanotubes, Electronic Structure, Molecular Dynamics, Raman
@article{Andrade2015,
title = {Linear Carbon Chains Under High Pressure Conditions},
author = {Nadia Ferreira Andrade, Acrísio L Aguiar, Yoong Ahm Kim, Morinobu Endo, Paulo TC Freire, Gustavo Bruneto, Douglas Soares Galvao, Mildred S Dresselhaus, Antonio Gomes Souza Filho},
url = {http://pubs.acs.org/doi/abs/10.1021/acs.jpcc.5b00902},
doi = {10.1021/acs.jpcc.5b00902},
year = {2015},
date = {2015-04-23},
journal = {The Journal of Physical Chemistry C},
volume = {119},
number = {19},
pages = {10669–10676},
abstract = {A high-pressure resonance Raman spectroscopy study of linear carbon chains encapsulated inside multiwalled carbon nanotubes (MWCNTs) is reported. While the frequencies of the tangential modes of carbon nanotubes (G band) harden as the pressure increases, the vibrational frequencies of the chain modes (around 1850 cm–1) decrease, thus indicating a softening of the carbon–carbon bonds in this 1D solid. Pressure-induced irreversible structural changes in the linear carbon chains are unveiled by the red shift in the vibrational modes when pressure is released. These results have been interpreted as being due to a coalescence of carbon chains, and this hypothesis is supported by state-of-the-art atomistic reactive molecular dynamics simulations.},
keywords = {Atomic Chains, Carbon Nanotubes, Electronic Structure, Molecular Dynamics, Raman},
pubstate = {published},
tppubtype = {article}
}
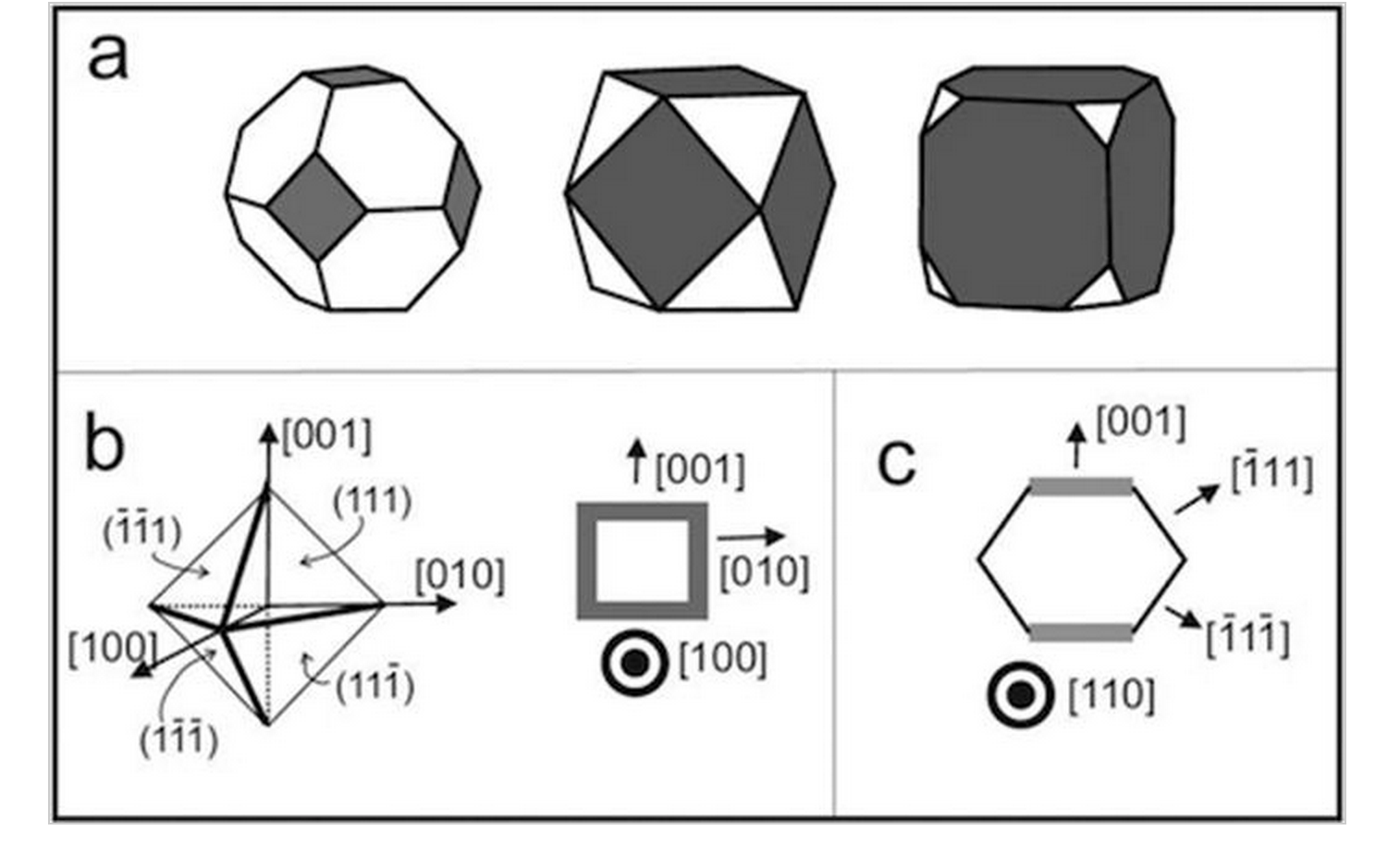
PAS Autreto MJ Lagos, J Bettini
Surface effects on the mechanical elongation of AuCu nanowires: De-alloying and the formation of mixed suspended atomic chains Journal Article
In: Journal of Applied Physics, vol. 117, no. 9, pp. 094301, 2015.
Abstract | Links | BibTeX | Tags: Metallic Nanowires, Molecular Dynamics, TEM, Theory of Electronic Indices
@article{Lagos2015,
title = {Surface effects on the mechanical elongation of AuCu nanowires: De-alloying and the formation of mixed suspended atomic chains},
author = {MJ Lagos, PAS Autreto, J Bettini, F Sato, SO Dantas, DS Galvao, D Ugarte},
url = {http://scitation.aip.org/content/aip/journal/jap/117/9/10.1063/1.4913625},
doi = {10.1063/1.4913625},
year = {2015},
date = {2015-03-07},
journal = {Journal of Applied Physics},
volume = {117},
number = {9},
pages = {094301},
abstract = {We report here an atomistic study of the mechanical deformation of Au x Cu (1− x ) atomic-size wires (nanowires (NWs)) by means of high resolution transmission electron microscopy experiments. Molecular dynamics simulations were also carried out in order to obtain deeper insights on the dynamical properties of stretched NWs. The mechanical properties are significantly dependent on the chemical composition that evolves in time at the junction; some structures exhibit a remarkable de-alloying behavior. Also, our results represent the first experimental realization of mixed linear atomic chains (LACs) among transition and noble metals; in particular, surface energies induce chemical gradients on NW surfaces that can be exploited to control the relative LAC compositions (different number of gold and copper atoms). The implications of these results for nanocatalysis and spin transport of one-atom-thick metal wires are addressed.},
keywords = {Metallic Nanowires, Molecular Dynamics, TEM, Theory of Electronic Indices},
pubstate = {published},
tppubtype = {article}
}
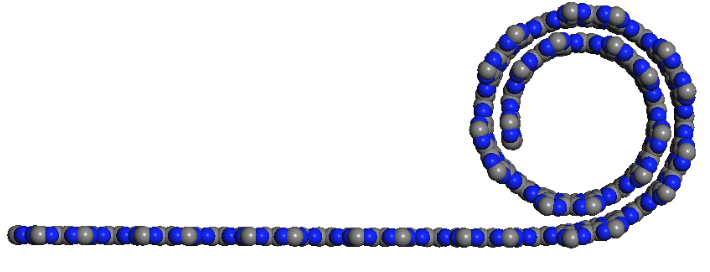
Eric Perim, Douglas S. Galvao
Novel Nanoscroll Structures from Carbon Nitride Layers Online
2015, (ArXiv Draft MRS Proceedings, 1726, mrsf14-1726-j05-02 (2015)).
Abstract | Links | BibTeX | Tags: carbon nitride, Molecular Dynamics, Scrolls
@online{Perim2015,
title = {Novel Nanoscroll Structures from Carbon Nitride Layers},
author = {Eric Perim, Douglas S. Galvao},
url = {http://arxiv.org/abs/1502.00260},
year = {2015},
date = {2015-02-02},
abstract = {Nanoscrolls consist of sheets rolled up into a papyrus-like form. Their open ends produce great radial flexibility, which can be exploited for a large variety of applications, from actuators to hydrogen storage. They have been successfully synthesized from different materials, including carbon and boron nitride. In this work we have investigated, through fully atomistic molecular dynamics simulations, the dynamics of scroll formation for a series of graphene-like carbon nitride (CN) two-dimensional systems: g-CN, triazine-based (g-C3N4), and heptazine-based (g-C3N4). Carbon nitride (CN) structures have been attracting great attention since their prediction as super hard materials. Recently, graphene-like carbon nitride (g-CN) structures have been synthesized with distinct stoichiometry and morphologies. By combining these unique CN characteristics with the structural properties inherent to nanoscrolls new nanostructures with very attractive mechanical and electronic properties could be formed. Our results show that stable nanoscrolls can be formed for all of CN structures we have investigated here. As the CN sheets have been already synthesized, these new scrolled structures are perfectly feasible and within our present-day technology.},
note = {ArXiv Draft MRS Proceedings, 1726, mrsf14-1726-j05-02 (2015)},
keywords = {carbon nitride, Molecular Dynamics, Scrolls},
pubstate = {published},
tppubtype = {online}
}
L. D. Machado E. Perim, D. S. Galvao
A Brief Review on Syntheses, Structures and Applications of Nanoscrolls Online
2015, (ArXiv draft of Frontiers in Materials, 1 pp. 31, 2014).
Abstract | Links | BibTeX | Tags: Molecular Dynamics, Scrolls
@online{Perim2015b,
title = {A Brief Review on Syntheses, Structures and Applications of Nanoscrolls},
author = {E. Perim, L. D. Machado, D. S. Galvao},
url = {http://arxiv.org/abs/1501.05711 },
year = {2015},
date = {2015-01-21},
journal = {arXiv preprint 1501.05711v1},
abstract = {Nanoscrolls are papyrus-like nanostructures which present unique properties due to their open ended morphology. These properties can be exploited in a plethora of technological applications, leading to the design of novel and interesting devices. During the past decade, significant advances in the synthesis and characterization of these structures have been made, but many challenges still remain. In this mini review we provide an overview on their history, experimental synthesis methods, basic properties and application perspectives.},
note = {ArXiv draft of Frontiers in Materials, 1 pp. 31, 2014},
keywords = {Molecular Dynamics, Scrolls},
pubstate = {published},
tppubtype = {online}
}
Pedro A. S. Autreto, Douglas S. Galvao
Site dependent hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation Online
2015, (ArXiv draft of MRS Proceedings, 1726, mrsf14-1726-j02-02 (2015)).
Abstract | Links | BibTeX | Tags: Graphynes, Hydrogenation, Molecular Dynamics
@online{Autreto2015,
title = {Site dependent hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Pedro A. S. Autreto, Douglas S. Galvao},
url = {http://arxiv.org/abs/1501.04521},
year = {2015},
date = {2015-01-19},
journal = {arXiv preprint 1501.04521},
abstract = {Graphyne is a generic name for a carbon allotrope family of 2D structures, where acetylenic groups connect benzenoid rings, with the coexistence of sp and sp2 hybridized carbon atoms. In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics and structural changes of the hydrogenation of ALPHA, BETA, GAMMA graphyne forms. Our results showed that the existence of different sites for hydrogen bonding, related to single and triple bonds, makes the process of incorporating hydrogen atoms into graphyne membranes much more complex than the graphene ones. Our results also show that hydrogenation reactions are strongly site dependent and that the sp-hybridized carbon atoms are the preferential sites to chemical attacks. In our cases, the effectiveness of the hydrogenation (estimated from the number of hydrogen atoms covalently bonded to carbon atoms) follows the ALPHA, BETA, GAMMA graphyne structure ordering.},
note = {ArXiv draft of MRS Proceedings, 1726, mrsf14-1726-j02-02 (2015)},
keywords = {Graphynes, Hydrogenation, Molecular Dynamics},
pubstate = {published},
tppubtype = {online}
}

Leonardo D. Machado Cristiano F. Woellner, Pedro A. S. Autreto
The Influence of Morphology on the Charge Transport in Two-Phase Disordered Organic Systems Online
2015, (ArXiv draft of MRS Proceedings, 1737, mrsf14-1737-u18-21 (2015)).
Abstract | Links | BibTeX | Tags: Conducting Polymers, Monte Carlo, Transport
@online{Woellner2015,
title = {The Influence of Morphology on the Charge Transport in Two-Phase Disordered Organic Systems},
author = {Cristiano F. Woellner, Leonardo D. Machado, Pedro A. S. Autreto, Jose A. Freire, Douglas S. Galvao},
url = {http://arxiv.org/abs/1501.01343},
year = {2015},
date = {2015-01-01},
booktitle = {MRS Proceedings},
volume = {1737},
pages = {mrsf14-1737-u18-21},
abstract = {In this work we use a three-dimensional Pauli master equation to investigate the charge carrier mobility of a two-phase system, which can mimic donor-acceptor and amorphous- crystalline bulk heterojunctions. Our approach can be separated into two parts: the morphology generation and the charge transport modeling in the generated blend. The morphology part is based on a Monte Carlo simulation of binary mixtures (donor/acceptor). The second part is carried out by numerically solving the steady-state Pauli master equation. By taking the energetic disorder of each phase, their energy offset and domain morphology into consideration, we show that the carrier mobility can have a significant different behavior when compared to a one-phase system. When the energy offset is non-zero, we show that the mobility electric field dependence switches from negative to positive at a threshold field proportional to the energy offset. Additionally, the influence of morphology, through the domain size and the interfacial roughness parameters, on the transport was also investigated.},
note = {ArXiv draft of MRS Proceedings, 1737, mrsf14-1737-u18-21 (2015)},
keywords = {Conducting Polymers, Monte Carlo, Transport},
pubstate = {published},
tppubtype = {online}
}

Leonardo D Machado Cristiano F Woellner, Pedro AS Autreto
The Influence of Morphology on the Charge Transport in Two-Phase Disordered Organic Systems Proceedings
vol. 1737, no. mrsf14-1737-u18-21, 2015, (MRS Proceedings, 1737, mrsf14-1737-u18-21).
Abstract | Links | BibTeX | Tags: Conducting Polymers, Monte Carlo, Solar Cells
@proceedings{Woellner2015b,
title = {The Influence of Morphology on the Charge Transport in Two-Phase Disordered Organic Systems},
author = {Cristiano F Woellner, Leonardo D Machado, Pedro AS Autreto, José A Freire, Douglas S Galvão},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=9707375&fileId=S1946427415005023},
doi = {10.1557/opl.2015.502},
year = {2015},
date = {2015-01-01},
booktitle = {MRS Proceedings},
volume = {1737},
number = {mrsf14-1737-u18-21},
pages = {mrsf14-1737-u18-21},
abstract = {In this work we use a three-dimensional Pauli master equation to investigate the charge carrier mobility of a two-phase system, which can mimic donor-acceptor and amorphous-crystalline bulk heterojunctions. Our approach can be separated into two parts: the morphology generation and the charge transport modeling in the generated blend. The morphology part is based on a Monte Carlo simulation of binary mixtures (donor/acceptor). The second part is carried out by numerically solving the steady-state Pauli master equation. By taking the energetic disorder of each phase, their energy offset and domain morphology into consideration, we show that the carrier mobility can have a significant different behavior when compared to a one-phase system. When the energy offset is non-zero, we show that the mobility electric field dependence switches from negative to positive at a threshold field proportional to the energy offset. Additionally, the influence of morphology, through the domain size and the interfacial roughness parameters, on the transport was also investigated.
},
note = {MRS Proceedings, 1737, mrsf14-1737-u18-21},
keywords = {Conducting Polymers, Monte Carlo, Solar Cells},
pubstate = {published},
tppubtype = {proceedings}
}
Pedro A. S. Autreto, Douglas S. Galvao
Site Dependent Hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation Proceedings
vol. 1726, no. mrsf14-1726-j02-02, 2015, (MRS Proceedings, 1726, mrsf14-1726-j02-02 ).
Abstract | Links | BibTeX | Tags: Graphynes, Hydrogenation, Molecular Dynamics
@proceedings{Autreto2015b,
title = {Site Dependent Hydrogenation in Graphynes: A Fully Atomistic Molecular Dynamics Investigation},
author = {Pedro A. S. Autreto, Douglas S. Galvao},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=9702693&fulltextType=RA&fileId=S1946427415004649},
doi = {10.1557/opl.2015.464},
year = {2015},
date = {2015-01-01},
journal = {Mater. Res. Soc. Symp. Proc. },
volume = {1726},
number = {mrsf14-1726-j02-02},
abstract = {Graphyne is a generic name for a carbon allotrope family of 2D structures, where acetylenic groups connect benzenoid rings, with the coexistence of sp and sp2 hybridized carbon atoms. In this work we have investigated, through fully atomistic reactive molecular dynamics simulations, the dynamics and structural changes of the hydrogenation of α, β, and γ graphyne forms. Our results showed that the existence of different sites for hydrogen bonding, related to single and triple bonds, makes the process of incorporating hydrogen atoms into graphyne membranes much more complex than the graphene ones. Our results also show that hydrogenation reactions are strongly site dependent and that the sp-hybridized carbon atoms are the preferential sites to chemical attacks. In our cases, the effectiveness of the hydrogenation (estimated from the number of hydrogen atoms covalently bonded to carbon atoms) follows the α, β, γ-graphyne structure ordering.},
note = {MRS Proceedings, 1726, mrsf14-1726-j02-02 },
keywords = {Graphynes, Hydrogenation, Molecular Dynamics},
pubstate = {published},
tppubtype = {proceedings}
}

Eric Perim, Douglas S. Galvao
Novel Nanoscroll Structures from Carbon Nitride Layers Proceedings
vol. 1726, no. mrsf14-1726-j05-02, 2015, (MRS Proceedings, 1726, mrsf14-1726-j05-02 ).
Abstract | Links | BibTeX | Tags: carbon nitride, Molecular Dynamics, nanoscrolls
@proceedings{Perim2015b,
title = {Novel Nanoscroll Structures from Carbon Nitride Layers},
author = {Eric Perim, Douglas S. Galvao},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=9700860&fileId=S1946427415004650},
doi = {DOI: 10.1557/opl.2015.465},
year = {2015},
date = {2015-01-01},
volume = {1726},
number = {mrsf14-1726-j05-02},
abstract = {Nanoscrolls consist of sheets rolled up into a papyrus-like form. Their open ends produce great radial flexibility, which can be exploited for a large variety of applications, from actuators to hydrogen storage. They have been successfully synthesized from different materials, including carbon and boron nitride. In this work we have investigated, through fully atomistic molecular dynamics simulations, the dynamics of scroll formation for a series of graphene-like carbon nitride (CN) two-dimensional systems: g-CN, triazine-based (g-C3N4), and heptazine-based (g-C3N4). Carbon nitride (CN) structures have been attracting great attention since their prediction as super hard materials. Recently, graphene-like carbon nitride (g-CN) structures have been synthesized with distinct stoichiometry and morphologies. By combining these unique CN characteristics with the structural properties inherent to nanoscrolls new nanostructures with very attractive mechanical and electronic properties could be formed. Our results show that stable nanoscrolls can be formed for all of CN structures we have investigated here. As the CN sheets have been already synthesized, these new scrolled structures are perfectly feasible and within our present-day technology.},
note = {MRS Proceedings, 1726, mrsf14-1726-j05-02 },
keywords = {carbon nitride, Molecular Dynamics, nanoscrolls},
pubstate = {published},
tppubtype = {proceedings}
}

Nadia F. Andradea Gustavo Brunettoa, Douglas S. Galvao
High Pressure Induced Binding Between Linear Carbon Chains and Nanotubes Proceedings
vol. 1752, no. 53-58, 2015, (MRS Proceedings, 1752, pp 53-58).
Abstract | Links | BibTeX | Tags: CNT encapsulation, Electronic Structure, Linear Chains, Molecular Dynamics
@proceedings{Brunettoa2015,
title = {High Pressure Induced Binding Between Linear Carbon Chains and Nanotubes},
author = {Gustavo Brunettoa, Nadia F. Andradea, Douglas S. Galvao, Antonio G. Souza Filho},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=9553206&fileId=S1946427415000913},
doi = {10.1557/opl.2015.91},
year = {2015},
date = {2015-01-01},
volume = {1752},
number = {53-58},
abstract = {Recent studies of single-walled carbon nanotubes (CNTs) in aqueous media have showed that water can significantly affect the tube mechanical properties. CNTs under hydrostatic compression can preserve their elastic properties up to large pressure values, while exhibiting exceptional resistance to mechanical loadings. It was experimentally observed that CNTs with encapsulated linear carbon chains (LCCs), when subjected to high hydrostatic pressure values, present irreversible red shifts in some of their vibrational frequencies. In order to address the cause of this phenomenon, we have carried out fully atomistic reactive (ReaxFF) molecular dynamics (MD) simulations for model structures mimicking the experimental conditions. We have considered the cases of finite and infinite (cyclic boundary conditions) CNTs filled with LCCs (LCC@CNTs) of different lengths (from 9 up to 40 atoms). Our results show that increasing the hydrostatic pressure causes the CNT to be deformed in an inhomogeneous way due to the LCC presence. The LCC/CNT interface regions exhibit convex curvatures, which results in more reactive sites, thus favoring the formation of covalent chemical bonds between the chain and the nanotube. This process is irreversible with the newly formed bonds continuing to exist even after releasing the external pressure and causing an irreversibly red shift in the chain vibrational modes from 1850 to 1500 cm−1.},
note = {MRS Proceedings, 1752, pp 53-58},
keywords = {CNT encapsulation, Electronic Structure, Linear Chains, Molecular Dynamics},
pubstate = {published},
tppubtype = {proceedings}
}
2014
Perim, Eric; Machado, Leonardo Dantas; Galvao, Douglas Soares
A Brief Review on Syntheses, Structures, and Applications of Nanoscrolls Journal Article
In: Frontiers in Materials, vol. 1, pp. 31, 2014, (Invited Review Paper).
Abstract | Links | BibTeX | Tags: Molecular Dynamics, Scrolls
@article{perim2014brief,
title = {A Brief Review on Syntheses, Structures, and Applications of Nanoscrolls},
author = {Perim, Eric and Machado, Leonardo Dantas and Galvao, Douglas Soares},
url = {http://journal.frontiersin.org/Journal/10.3389/fmats.2014.00031/abstract},
year = {2014},
date = {2014-12-01},
journal = {Frontiers in Materials},
volume = {1},
pages = {31},
publisher = {Frontiers},
abstract = {Nanoscrolls are papyrus-like nanostructures, which present unique properties due to their open ended morphology. These properties can be exploited in a plethora of technological applications, leading to the design of novel and interesting devices. During the past decade, significant advances in the synthesis and characterization of these structures have been made, but many challenges still remain. In this mini review, we provide an overview on their history, experimental synthesis methods, basic properties, and application perspectives.},
note = {Invited Review Paper},
keywords = {Molecular Dynamics, Scrolls},
pubstate = {published},
tppubtype = {article}
}
Perim, E; Paupitz, R; Botari, T; Galvao, DS
One-dimensional silicon and germanium nanostructures with no carbon analogues Journal Article
In: Physical Chemistry Chemical Physics, vol. 16, no. 44, pp. 24570–24574, 2014.
Abstract | Links | BibTeX | Tags: DFT, Germanium, Nanotubes, Silicon
@article{perim2014one,
title = {One-dimensional silicon and germanium nanostructures with no carbon analogues},
author = {Perim, E and Paupitz, R and Botari, T and Galvao, DS},
url = {http://pubs.rsc.org/en/content/articlehtml/2014/cp/c4cp03708a},
year = {2014},
date = {2014-01-01},
journal = {Physical Chemistry Chemical Physics},
volume = {16},
number = {44},
pages = {24570--24574},
publisher = {Royal Society of Chemistry},
abstract = {In this work we report new silicon and germanium tubular nanostructures with no corresponding stable carbon analogues. The electronic and mechanical properties of these new tubes were investigated through ab initio methods. Our results show that these structures have lower energy than their corresponding nanoribbon structures and are stable up to high temperatures (500 and 1000 K, for silicon and germanium tubes, respectively). Both tubes are semiconducting with small indirect band gaps, which can be significantly altered by both compressive and tensile strains. Large bandgap variations of almost 50% were observed for strain rates as small as 3%, suggesting their possible applications in sensor devices. They also present high Young's modulus values (0.25 and 0.15 TPa, respectively). TEM images were simulated to help in the identification of these new structures.},
keywords = {DFT, Germanium, Nanotubes, Silicon},
pubstate = {published},
tppubtype = {article}
}
Botari, T; Perim, E; Autreto, PAS; van Duin, ACT; Paupitz, R; Galvao, DS
Mechanical properties and fracture dynamics of silicene membranes Journal Article
In: PHYSICAL CHEMISTRY CHEMICAL PHYSICS, vol. 16, no. 36, pp. 19417–19423, 2014.
Abstract | Links | BibTeX | Tags: Fracture, Germanene, Graphene, Mechanical Properties, Silicene
@article{botari2014mechanical,
title = {Mechanical properties and fracture dynamics of silicene membranes},
author = {Botari, T and Perim, E and Autreto, PAS and van Duin, ACT and Paupitz, R and Galvao, DS},
url = {http://pubs.rsc.org/en/content/articlehtml/2014/cp/c4cp02902j},
year = {2014},
date = {2014-01-01},
journal = {PHYSICAL CHEMISTRY CHEMICAL PHYSICS},
volume = {16},
number = {36},
pages = {19417--19423},
publisher = {ROYAL SOC CHEMISTRY},
abstract = {As graphene has become one of the most important materials, there is renewed interest in other similar structures. One example is silicene, the silicon analogue of graphene. It shares some of the remarkable graphene properties, such as the Dirac cone, but presents some distinct ones, such as a pronounced structural buckling. We have investigated, through density functional based tight-binding (DFTB), as well as reactive molecular dynamics (using ReaxFF), the mechanical properties of suspended single-layer silicene. We calculated the elastic constants, analyzed the fracture patterns and edge reconstructions. We also addressed the stress distributions, unbuckling mechanisms and the fracture dependence on the temperature. We analysed the differences due to distinct edge morphologies, namely zigzag and armchair.},
keywords = {Fracture, Germanene, Graphene, Mechanical Properties, Silicene},
pubstate = {published},
tppubtype = {article}
}
Brunetto, Gustavo; Andrade, Nadia F.; Galvao, Douglas S; Antonio Filho, G Souza
High Pressure Induced Binding Between Linear Carbon Chains and Nanotubes Proceedings
2014.
Abstract | Links | BibTeX | Tags: Atomic Chains, Carbon Nanotubes, Molecular Dynamics
@proceedings{brunetto2014high,
title = {High Pressure Induced Binding Between Linear Carbon Chains and Nanotubes},
author = {Brunetto, Gustavo and Andrade, Nadia F. and Galvao, Douglas S and Antonio Filho, G Souza},
url = {http://arxiv.org/abs/1412.7966},
year = {2014},
date = {2014-01-01},
journal = {arXiv preprint arXiv:1412.7966},
abstract = {Recent studies of single-walled carbon nanotubes (CNTs) in aqueous media have showed that water can significantly affect the tube mechanical properties. CNTs under hydrostatic compression can preserve their elastic properties up to large pressure values, while exhibiting exceptional resistance to mechanical loadings. It was experimentally observed that CNTs with encapsulated linear carbon chains (LCCs), when subjected to high hydrostatic pressure values, present irreversible red shifts in some of their vibrational frequencies. In order to address the cause of this phenomenon, we have carried out fully atomistic reactive (ReaxFF) molecular dynamics (MD) simulations for model structures mimicking the experimental conditions. We have considered the cases of finite and infinite (cyclic boundary conditions) CNTs filled with LCCs (LCC inside CNTs) of different lengths (from 9 up to 40 atoms). Our results show that increasing the hydrostatic pressure causes the CNT to be deformed in an inhomogeneous way due to the LCC presence. The LCC-CNT interface regions exhibit convex curvatures, which results in more reactive sites, thus favoring the formation of covalent chemical bonds between the chain and the nanotube. This process is irreversible with the newly formed bonds continuing to exist even after releasing the external pressure and causing an irreversibly red shift in the chain vibrational modes from 1850 to 1500 cm−1.},
keywords = {Atomic Chains, Carbon Nanotubes, Molecular Dynamics},
pubstate = {published},
tppubtype = {proceedings}
}
da Silva Autreto, Pedro Alves; Galvao, Douglas S.; Artacho, Emilio
Species fractionation in atomic chains from mechanically stretched alloys Journal Article
In: JOURNAL OF PHYSICS-CONDENSED MATTER, vol. 26, no. 43, 2014, ISSN: 0953-8984.
@article{daAutreto2014,
title = {Species fractionation in atomic chains from mechanically stretched alloys},
author = {da Silva Autreto, Pedro Alves and Galvao, Douglas S. and Artacho, Emilio},
issn = {0953-8984},
year = {2014},
date = {2014-01-01},
journal = {JOURNAL OF PHYSICS-CONDENSED MATTER},
volume = {26},
number = {43},
abstract = {Bettini et al (2006 Nat. Nanotechnol. 1 182-5) reported the first experimental realization of linear atomic chains (LACs) composed of different atoms (Au and Ag). The different contents of Au and Ag were observed in the chains from what was found in the bulk alloys, which raises the question of what the wire composition is, if it is in equilibrium with a bulk alloy. In this work we address the thermodynamic driving force for species fractionation in LACs under tension, and we present the density-functional theory results for Ag-Au chain alloys. A pronounced stabilization of the wires with an alternating Ag-Au sequence is observed, which could be behind the experimentally observed Au enrichment in LACs from alloys with high Ag content.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Gao, Guanhui; Mathkar, Akshay; Martins, Eric Perim; Galvao, Douglas S; Gao, Duyang; da Silva Autreto, Pedro Alves; Sun, Chengjun; Cai, Lintao; Ajayan, Pulickel M
Designing nanoscaled hybrids from atomic layered boron nitride with silver nanoparticle deposition Journal Article
In: Journal of Materials Chemistry A, vol. 2, no. 9, pp. 3148–3154, 2014.
Abstract | Links | BibTeX | Tags: DFT, nano particles
@article{gao2014designing,
title = {Designing nanoscaled hybrids from atomic layered boron nitride with silver nanoparticle deposition},
author = {Gao, Guanhui and Mathkar, Akshay and Martins, Eric Perim and Galvao, Douglas S and Gao, Duyang and da Silva Autreto, Pedro Alves and Sun, Chengjun and Cai, Lintao and Ajayan, Pulickel M},
url = {http://pubs.rsc.org/en/Content/ArticleLanding/2014/TA/c3ta12892j#!divAbstract},
year = {2014},
date = {2014-01-01},
journal = {Journal of Materials Chemistry A},
volume = {2},
number = {9},
pages = {3148--3154},
publisher = {Royal Society of Chemistry},
abstract = {We have developed a microwave assisted one-pot approach to fabricate a novel hybrid nano-composite composed of two-dimensional chemically exfoliated layered hexagonal boron nitride (h-BN) and embedded silver nanoparticles (SNP). Atomic layered h-BN exfoliated using chemical liquid showed strong in-plane bonding and weak van der Waals interplanar interactions, which is utilized for chemically interfacing SNP, indicating their ability to act as excellent nano-scaffolds. The SNP/h-BN optical response, in particular band gap, is strongly dependent on the concentration of the metallic particles. In order to gain further insight into this behavior we have also carried out ab initio density functional theory (DFT) calculations on modeled structures, demonstrating that the bandgap value of SNP/h-BN hybrids could be significantly altered by a small percentage of OH− groups located at dangling B and N atoms. Our results showed that these novel SNP/h-BN nanohybrid structures exhibited excellent thermal stability and they are expected to be applied as devices for thermal oxidation-resistant surface enhanced Raman spectroscopy (SERS). The SNP/h-BN membrane showed remarkable antibacterial activity, suggesting their potential use in water disinfection and food packaging.
},
keywords = {DFT, nano particles},
pubstate = {published},
tppubtype = {article}
}

Perim, E; Fonseca, AF; Pugno, NM; Galvao, DS
Violation of the universal behavior of membranes inside cylindrical tubes at nanoscale Journal Article
In: EPL (Europhysics Letters), vol. 105, no. 5, pp. 56002, 2014.
Abstract | Links | BibTeX | Tags: Graphene, Nanoscale Effects, Scrolls
@article{perim2014violation,
title = {Violation of the universal behavior of membranes inside cylindrical tubes at nanoscale},
author = {Perim, E and Fonseca, AF and Pugno, NM and Galvao, DS},
url = {http://iopscience.iop.org/0295-5075/105/5/56002},
year = {2014},
date = {2014-01-01},
journal = {EPL (Europhysics Letters)},
volume = {105},
number = {5},
pages = {56002},
publisher = {IOP Publishing},
abstract = {Recently, it was proposed based on classical elasticity theory and experiments at macroscale, that the conformations of sheets inside cylindrical tubes present a universal behavior. A natural question is whether this behavior still holds at nanoscale. Based on molecular-dynamics simulations and analytical modeling for graphene and boron nitride membranes confined inside carbon nanotubes, we show that the class of universality observed at macroscale is violated at nanoscale. The precise origin of these discrepancies is addressed and proven to be related to both surface and atomistic effects.
},
keywords = {Graphene, Nanoscale Effects, Scrolls},
pubstate = {published},
tppubtype = {article}
}
Perim, Eric; Paupitz, Ricardo; Botari, Tiago; Galvao, Douglas S
Novel Semiconducting Silicon and Germanium Nanotubes Journal Article
In: arXiv preprint arXiv:1403.2061, 2014.
Abstract | Links | BibTeX | Tags: DFT, Germanium, Nanotubes, Silicon
@article{perim2014novel,
title = {Novel Semiconducting Silicon and Germanium Nanotubes},
author = {Perim, Eric and Paupitz, Ricardo and Botari, Tiago and Galvao, Douglas S},
url = {http://arxiv.org/abs/1403.2061},
year = {2014},
date = {2014-01-01},
journal = {arXiv preprint arXiv:1403.2061},
abstract = {In this work we report new silicon and germanium nanotube structures, with no corresponding
stable carbon analogues and which cannot be described by integer chiral indices. The electronic
and mechanical properties of these new tubes were investigated through ab initio methods. Our
results show that the structures are stable up to high temperatures (500 and 1000 K, for silicon and
germanium tubes, respectively). Both tubes are semiconducting with small indirect band gaps,
which can be significantly altered by both compressive and tensile strains. They also present high
Young modulus values (0.25 and 0.15 TPa, respectively).},
keywords = {DFT, Germanium, Nanotubes, Silicon},
pubstate = {published},
tppubtype = {article}
}
stable carbon analogues and which cannot be described by integer chiral indices. The electronic
and mechanical properties of these new tubes were investigated through ab initio methods. Our
results show that the structures are stable up to high temperatures (500 and 1000 K, for silicon and
germanium tubes, respectively). Both tubes are semiconducting with small indirect band gaps,
which can be significantly altered by both compressive and tensile strains. They also present high
Young modulus values (0.25 and 0.15 TPa, respectively).
Brunetto, G; Galvao, DS
Graphene-like Membranes: From Impermeable to Selective Sieves Proceedings
Cambridge University Press, vol. 1658, 2014.
Abstract | Links | BibTeX | Tags: Graphene, Membranes, Porous Graphene, Sieves
@proceedings{brunetto2014graphene,
title = {Graphene-like Membranes: From Impermeable to Selective Sieves},
author = {Brunetto, G and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=9248039&fileId=S1946427414004011},
year = {2014},
date = {2014-01-01},
journal = {MRS Proceedings},
volume = {1658},
pages = {mrsf13--1658},
publisher = {Cambridge University Press},
abstract = {Recently, it was proposed that graphene membranes could act as impermeable atomic
structures to standard gases. For some other applications, a higher level of porosity is needed,
and the so-called Porous Graphene (PG) and Biphenylene Carbon (BPC) membranes are good
candidates to effectively work as selective sieves. In this work we have used classical molecular
dynamics simulations to study the dynamics of membrane permeation of He and Ar atoms and
possible selectivity effects. For the graphene membranes we did not observe any leakage
through the membrane and/or membrane/substrate interface until a critical pressure limit, then a
sudden membrane detachment occurs. PG and BPC membranes are not impermeable as
graphene ones, but there are significant energy barriers to diffusion depending on the atom type.
Our results show that this kind of porous membranes can be effectively used as selective sieves
for pure and mixtures of gases.},
keywords = {Graphene, Membranes, Porous Graphene, Sieves},
pubstate = {published},
tppubtype = {proceedings}
}
structures to standard gases. For some other applications, a higher level of porosity is needed,
and the so-called Porous Graphene (PG) and Biphenylene Carbon (BPC) membranes are good
candidates to effectively work as selective sieves. In this work we have used classical molecular
dynamics simulations to study the dynamics of membrane permeation of He and Ar atoms and
possible selectivity effects. For the graphene membranes we did not observe any leakage
through the membrane and/or membrane/substrate interface until a critical pressure limit, then a
sudden membrane detachment occurs. PG and BPC membranes are not impermeable as
graphene ones, but there are significant energy barriers to diffusion depending on the atom type.
Our results show that this kind of porous membranes can be effectively used as selective sieves
for pure and mixtures of gases.
Bizao, RA; Botari, T; Galvao, DS
Mechanical Properties of Graphene Nanowiggles Proceedings
Cambridge University Press, vol. 1658, 2014.
Abstract | Links | BibTeX | Tags: Graphene, Molecular Dynamics, NanoRibbons, Nanowiggles
@proceedings{bizao2014mechanical,
title = {Mechanical Properties of Graphene Nanowiggles},
author = {Bizao, RA and Botari, T and Galvao, DS},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=9248042&fileId=S1946427414004023},
year = {2014},
date = {2014-01-01},
journal = {MRS Proceedings},
volume = {1658},
pages = {mrsf13--1658},
publisher = {Cambridge University Press},
abstract = {In this work we have investigated the mechanical properties and fracture patterns of some graphene nanowiggles (GNWs). Graphene nanoribbons are finite graphene segments with a large aspect ratio, while GNWs are nonaligned periodic repetitions of graphene nanoribbons. We have carried out fully atomistic molecular dynamics simulations using a reactive force field (ReaxFF), as implemented in the LAMPPS (Large-scale Atomic/Molecular Massively Parallel Simulator) code. Our results showed that the GNW fracture patterns are strongly dependent on the nanoribbon topology and present an interesting behavior, since some narrow sheets have larger ultimate failure strain values. This can be explained by the fact that narrow nanoribbons have more angular freedom when compared to wider ones, which can create a more efficient way to accumulate and to dissipate strain/stress. We have also observed the formation of linear atomic chains (LACs) and some structural defect reconstructions during the material rupture. The reported graphene failure patterns, where zigzag/armchair edge terminated graphene structures are fractured along armchair/zigzag lines, were not observed in the GNW analyzed cases.},
keywords = {Graphene, Molecular Dynamics, NanoRibbons, Nanowiggles},
pubstate = {published},
tppubtype = {proceedings}
}
Perim, Eric; Galvao, Douglas S
Novel Nanoscroll Structures from Carbon Nitride Layers Journal Article
In: ChemPhysChem, vol. 15, no. 11, pp. 2367–2371, 2014.
Abstract | Links | BibTeX | Tags: carbon nitride, Molecular Dynamics, Scrolls
@article{perim2014novelb,
title = {Novel Nanoscroll Structures from Carbon Nitride Layers},
author = {Perim, Eric and Galvao, Douglas S},
url = {http://onlinelibrary.wiley.com/doi/10.1002/cphc.201402059/full},
year = {2014},
date = {2014-01-01},
journal = {ChemPhysChem},
volume = {15},
number = {11},
pages = {2367--2371},
publisher = {WILEY-VCH Verlag},
abstract = {Nanoscrolls (papyrus-like nanostructures) are very attractive structures for a variety of applications, owing to their tunable diameter and large accessible surface area. They have been successfully synthesized from different materials. In this work, we investigate, through fully atomistic molecular dynamics simulations, the dynamics of scroll formation for a series of graphene-like carbon nitride (CN) two-dimensional systems: g-CN, triazine-based g-C3N4, and heptazine-based g-C3N4. Our results show that stable nanoscrolls can be formed for each of these structures. Possible synthetic routes to produce these nanostructures are also addressed.},
keywords = {carbon nitride, Molecular Dynamics, Scrolls},
pubstate = {published},
tppubtype = {article}
}
Autreto, PAS; de Sousa, JM; Galvao, DS
Site-dependent hydrogenation on graphdiyne Journal Article
In: Carbon, vol. 77, pp. 829–834, 2014.
Abstract | Links | BibTeX | Tags: Functionalization, Graphdyine, Graphene, Graphynes
@article{autreto2014site,
title = {Site-dependent hydrogenation on graphdiyne},
author = {Autreto, PAS and de Sousa, JM and Galvao, DS},
url = {http://www.sciencedirect.com/science/article/pii/S0008622314005429},
year = {2014},
date = {2014-01-01},
journal = {Carbon},
volume = {77},
pages = {829--834},
publisher = {Pergamon},
abstract = {Graphene is one of the most important materials in science today due to its unique and remarkable electronic, thermal and mechanical properties. However in its pristine state, graphene is a gapless semiconductor, what limits its use in transistor electronics. In part due to the revolution created by graphene in materials science, there is a renewed interest in other possible graphene-like two-dimensional structures. Examples of these structures are graphynes and graphdiynes, which are two-dimensional structures, composed of carbon atoms in sp2 and sp-hybridized states. Graphdiynes (benzenoid rings connecting two acetylenic groups) were recently synthesized and some of them are intrinsically nonzero gap systems. These systems can be easily hydrogenated and the relative level of hydrogenation can be used to tune the band gap values. We have investigated, using fully reactive molecular dynamics (ReaxFF), the structural and dynamics aspects of the hydrogenation mechanisms of graphdiyne membranes. Our results showed that the hydrogen bindings have different atom incorporation rates and that the hydrogenation patterns change in time in a very complex way. The formation of correlated domains reported to hydrogenated graphene is no longer observed in graphdiyne cases.},
keywords = {Functionalization, Graphdyine, Graphene, Graphynes},
pubstate = {published},
tppubtype = {article}
}
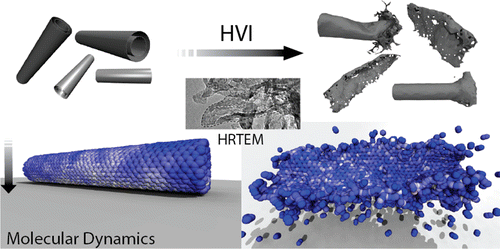
Ozden, Sehmus; Autreto, Pedro AS; Tiwary, Chandra Sekhar; Khatiwada, Suman; Machado, Leonardo; Galvao, Douglas S; Vajtai, Robert; Barrera, Enrique V; M. Ajayan, Pulickel
Unzipping Carbon Nanotubes at High Impact Journal Article
In: Nano letters, vol. 14, no. 7, pp. 4131–4137, 2014.
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, Fracture, Unzipping
@article{ozden2014unzipping,
title = {Unzipping Carbon Nanotubes at High Impact},
author = {Ozden, Sehmus and Autreto, Pedro AS and Tiwary, Chandra Sekhar and Khatiwada, Suman and Machado, Leonardo and Galvao, Douglas S and Vajtai, Robert and Barrera, Enrique V and M. Ajayan, Pulickel},
url = {http://pubs.acs.org/doi/abs/10.1021/nl501753n},
year = {2014},
date = {2014-01-01},
journal = {Nano letters},
volume = {14},
number = {7},
pages = {4131--4137},
publisher = {American Chemical Society},
abstract = {The way nanostructures behave and mechanically respond to high impact collision is a topic of intrigue. For anisotropic nanostructures, such as carbon nanotubes, this response will be complicated based on the impact geometry. Here we report the result of hypervelocity impact of nanotubes against solid targets and show that impact produces a large number of defects in the nanotubes, as well as rapid atom evaporation, leading to their unzipping along the nanotube axis. Fully atomistic reactive molecular dynamics simulations are used to gain further insights of the pathways and deformation and fracture mechanisms of nanotubes under high energy mechanical impact. Carbon nanotubes have been unzipped into graphene nanoribbons before using chemical treatments but here the instability of nanotubes against defect formation, fracture, and unzipping is revealed purely through mechanical impact.},
keywords = {Carbon Nanotubes, Fracture, Unzipping},
pubstate = {published},
tppubtype = {article}
}

Vinod, Soumya; Tiwary, Chandra Sekhar; da Silva Autreto, Pedro Alves; Taha-Tijerina, Jaime; Ozden, Sehmus; Chipara, Alin Cristian; Vajtai, Robert; Galvao, Douglas S; Narayanan, Tharangattu N; Ajayan, Pulickel M
Low-density three-dimensional foam using self-reinforced hybrid two-dimensional atomic layers Journal Article
In: Nature Communications, vol. 5, 2014.
Links | BibTeX | Tags: foams, Fracture, Graphene, Mechanical Properties, top20
@article{vinod2014low,
title = {Low-density three-dimensional foam using self-reinforced hybrid two-dimensional atomic layers},
author = {Vinod, Soumya and Tiwary, Chandra Sekhar and da Silva Autreto, Pedro Alves and Taha-Tijerina, Jaime and Ozden, Sehmus and Chipara, Alin Cristian and Vajtai, Robert and Galvao, Douglas S and Narayanan, Tharangattu N and Ajayan, Pulickel M},
url = {http://www.nature.com/ncomms/2014/140729/ncomms5541/full/ncomms5541.html},
year = {2014},
date = {2014-01-01},
journal = {Nature Communications},
volume = {5},
publisher = {Nature Publishing Group},
keywords = {foams, Fracture, Graphene, Mechanical Properties, top20},
pubstate = {published},
tppubtype = {article}
}
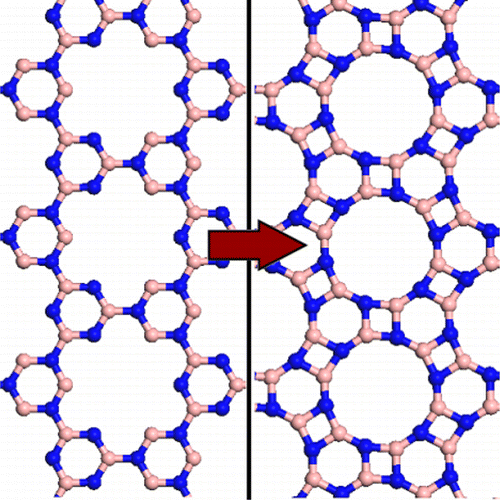
Perim, Eric; Paupitz, Ricardo; Autreto, PAS; Galvao, Douglas Soares
Inorganic Graphenylene: A Porous Two-Dimensional Material With Tunable Band Gap Journal Article
In: The Journal of Physical Chemistry C, vol. 118, no. 41, pp. 23670–23674, 2014.
Abstract | Links | BibTeX | Tags: BPC, Graphenylene, Porous Graphene
@article{perim2014inorganic,
title = {Inorganic Graphenylene: A Porous Two-Dimensional Material With Tunable Band Gap},
author = {Perim, Eric and Paupitz, Ricardo and Autreto, PAS and Galvao, Douglas Soares},
url = {http://pubs.acs.org/doi/abs/10.1021/jp502119y},
year = {2014},
date = {2014-01-01},
journal = {The Journal of Physical Chemistry C},
volume = {118},
number = {41},
pages = {23670--23674},
publisher = {American Chemical Society},
abstract = {By means of ab initio calculations, we investigate the possibility of existence of a boron nitride (BN) porous two-dimensional nanosheet, which is geometrically similar to the carbon allotrope known as biphenylene carbon. The proposed structure, which we called inorganic graphenylene (IGP), is formed spontaneously after selective dehydrogenation of the porous boron nitride (BN) structure proposed by Ding et al. We study the structural and electronic properties of both porous BN and IGP, and it is shown that, by selective substitution of B and N atoms with carbon atoms in these structures, the band gap can be significantly reduced, changing their behavior from insulators to semiconductors, thus opening the possibility of band gap engineering for this class of two-dimensional materials.},
keywords = {BPC, Graphenylene, Porous Graphene},
pubstate = {published},
tppubtype = {article}
}
2013
Paupitz, R; Autreto, Pedro AS; Legoas, SB; Srinivasan, S Goverapet; van Duin, Adri CT; Galvao, DS
Graphene to fluorographene and fluorographane: a theoretical study Journal Article
In: Nanotechnology, vol. 24, no. 3, pp. 035706, 2013.
Abstract | Links | BibTeX | Tags: Fluorographene, Graphane, Graphene
@article{paupitz2013graphene,
title = {Graphene to fluorographene and fluorographane: a theoretical study},
author = {Paupitz, R and Autreto, Pedro AS and Legoas, SB and Srinivasan, S Goverapet and van Duin, Adri CT and Galvao, DS},
url = {http://iopscience.iop.org/0957-4484/24/3/035706},
year = {2013},
date = {2013-01-01},
journal = {Nanotechnology},
volume = {24},
number = {3},
pages = {035706},
publisher = {IOP Publishing},
abstract = {We report here a fully reactive molecular dynamics study on the structural and dynamical aspects of the fluorination of graphene membranes (fluorographene). Our results show that fluorination tends to produce defective areas on the graphene membranes with significant distortions of carbon–carbon bonds. Depending on the amount of incorporated fluorine atoms, large membrane holes were observed due to carbon atom losses. These results may explain the broad distribution of the structural lattice parameter values experimentally observed. We have also investigated the effects of mixing hydrogen and fluorine atoms on the graphene functionalization. Our results show that, when in small amounts, the presence of hydrogen atoms produces a significant decrease in the rate of fluorine incorporation onto the membrane. On the other hand, when fluorine is the minority element, it produces a significant catalytic effect on the rate of hydrogen incorporation. We have also observed the spontaneous formation of new hybrid structures with different stable configurations (chair-like, zigzag-like and boat-like) which we named fluorographane.},
keywords = {Fluorographene, Graphane, Graphene},
pubstate = {published},
tppubtype = {article}
}

Perim, Eric; Paupitz, Ricardo; Galvao, Douglas S
Controlled route to the fabrication of carbon and boron nitride nanoscrolls: A molecular dynamics investigation Journal Article
In: Journal of Applied Physics, vol. 113, no. 5, pp. 054306, 2013.
Abstract | Links | BibTeX | Tags: Boron Nitride, Carbon Nanotubes, Graphene, Molecular Dynamics, Scrolls
@article{perim2013controlled,
title = {Controlled route to the fabrication of carbon and boron nitride nanoscrolls: A molecular dynamics investigation},
author = {Perim, Eric and Paupitz, Ricardo and Galvao, Douglas S},
url = {http://scitation.aip.org/content/aip/journal/jap/113/5/10.1063/1.4790304},
year = {2013},
date = {2013-01-01},
journal = {Journal of Applied Physics},
volume = {113},
number = {5},
pages = {054306},
publisher = {AIP Publishing},
abstract = {Carbon nanoscrolls (graphene layers rolled up into papyrus-like tubular structures) are nanostructures with unique and interesting characteristics that could be exploited to build several new nanodevices. However, an efficient and controlled synthesis of these structures was not achieved yet, making its large scale production a challenge to materials scientists. Also, the formation process and detailed mechanisms that occur during its synthesis are not completely known. In this work, using fully atomistic molecular dynamics simulations, we discuss a possible route to nanoscrolls made from graphene layers deposited over silicon oxide substrates containing chambers/pits. The scrolling mechanism is triggered by carbon nanotubes deposited on the layers. The process is completely general and can be used to produce scrolls from other lamellar materials, like boron nitride, for instance.},
keywords = {Boron Nitride, Carbon Nanotubes, Graphene, Molecular Dynamics, Scrolls},
pubstate = {published},
tppubtype = {article}
}

SB Legoas LD Machado, JS Soares
Dynamics of the formation of carbon nanotube serpentines Journal Article
In: Physical Review Letters, vol. 110, no. 10, pp. 105502, 2013.
Abstract | Links | BibTeX | Tags: Carbon Nanotubes, Molecular Dynamics, Serpentines, top20
@article{machado2013dynamics,
title = {Dynamics of the formation of carbon nanotube serpentines},
author = {LD Machado, SB Legoas, JS Soares, N Shadmi, A Jorio, E Joselevich, DS Galvão},
url = {http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.110.105502},
year = {2013},
date = {2013-01-01},
journal = {Physical Review Letters},
volume = {110},
number = {10},
pages = {105502},
publisher = {American Physical Society},
abstract = {Recently, Geblinger et al. [Nat. Nanotechnol. 3, 195 (2008)] reported the experimental realization of carbon nanotube S-like shaped nanostructures, the so-called carbon nanotube serpentines. We report here results from multimillion fully atomistic molecular dynamics simulations of their formation. We consider one-μm-long carbon nanotubes placed on stepped substrates with and without a catalyst nanoparticle on the top free end of the tube. A force is applied to the upper part of the tube during a short period of time and turned off; then the system is set free to evolve in time. Our results show that these conditions are sufficient to form robust serpentines and validates the general features of the “falling spaghetti model” proposed to explain their formation.
},
keywords = {Carbon Nanotubes, Molecular Dynamics, Serpentines, top20},
pubstate = {published},
tppubtype = {article}
}

Perim, Eric; Santos, Ricardo Paupitz; Autreto, Pedro Alves da Silva; Galvao, Douglas S
Fracture Patterns of Boron Nitride Nanotubes Proceedings
Cambridge University Press, vol. 1526, 2013.
Abstract | Links | BibTeX | Tags: Boron Nitride, Fracture, Mechanical Properties, Unzipping
@proceedings{perim2013fracture,
title = {Fracture Patterns of Boron Nitride Nanotubes},
author = {Perim, Eric and Santos, Ricardo Paupitz and Autreto, Pedro Alves da Silva and Galvao, Douglas S},
url = {http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=8883390&fileId=S1946427413004946},
year = {2013},
date = {2013-01-01},
journal = {MRS Proceedings},
volume = {1526},
pages = {mrsf12--1526},
publisher = {Cambridge University Press},
abstract = {During the last years carbon-based nanostructures (such as, fullerenes, carbon nanotubes and graphene) have been object of intense investigations. The great interest in these nanostructures can be attributed to their remarkable electrical and mechanical properties. Their inorganic equivalent structures do exist and are based on boron nitride (BN) motifs. BN fullerenes, nanotubes and single layers have been already synthesized. Recently, the fracture patterns of single layer graphene and multi-walled carbon nanotubes under stress have been studied by theoretical and experimental methods. In this work we investigated the fracturing process of defective carbon and boron nitride nanotubes under similar stress conditions. We have carried out fully atomistic molecular reactive molecular dynamics simulations using the ReaxFF force field. The similarities and differences between carbon and boron nitride fracture patterns are addressed.},
keywords = {Boron Nitride, Fracture, Mechanical Properties, Unzipping},
pubstate = {published},
tppubtype = {proceedings}
}
http://scholar.google.com/citations?hl=en&user=95SvbM8AAAAJ

